HOW PROTEINS ARE CONTROLLED
Thus far, we have examined how binding to other molecules allows proteins to perform their specific functions. But inside the cell, most proteins and enzymes do not work continuously, or at full speed. Instead, their activities are regulated in a coordinated fashion so the cell can maintain itself in an optimal state, producing only those molecules it requires to thrive under current conditions. By coordinating not only when—and how vigorously—proteins perform, but also where in the cell they act, the cell ensures that it does not deplete its energy reserves by accumulating molecules it does not need or waste its stockpiles of critical substrates. We now consider how cells control the activity of their enzymes and other proteins.
The regulation of protein activity occurs at many levels. At the most fundamental level, the cell controls the amount of each protein it contains. It can do so by controlling the expression of the gene that encodes that protein (discussed in Chapter 8). It can also regulate the rate at which the protein is degraded (discussed in Chapter 7). The cell also controls protein activities by confining the participating proteins to particular subcellular compartments. Some of these compartments are enclosed by membranes (as discussed in Chapters 11, 12, 14, and 15); others are created by the proteins that are drawn there, as we discuss shortly. Finally, the activity of an individual protein can be rapidly adjusted at the level of the protein itself.
All of these mechanisms rely on the ability of proteins to interact with other molecules—including other proteins. These interactions can cause proteins to adopt different conformations, and thereby alter their function, as we see next.
The Catalytic Activities of Enzymes Are Often Regulated by Other Molecules
A living cell contains thousands of different enzymes, many of which are operating at the same time in the same small volume of the cytosol. By their catalytic action, enzymes generate a complex web of metabolic pathways, each composed of chains of chemical reactions in which the product of one enzyme becomes the substrate of the next. In this maze of pathways, there are many branch points where different enzymes compete for the same substrate. The system is so complex that elaborate controls are required to regulate when and how rapidly each reaction occurs.
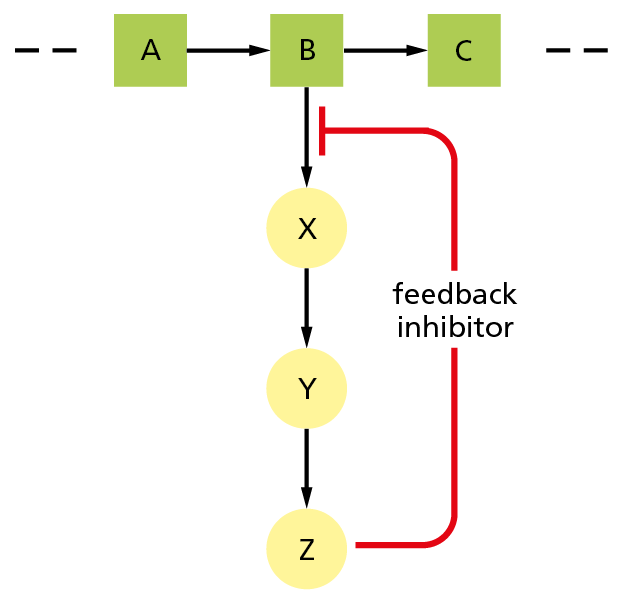
Figure 4−42 Feedback inhibition regulates the flow through biosynthetic pathways. B is the first metabolite in a pathway that gives the end product Z. Z inhibits the first enzyme that is specific to its own synthesis and thereby limits its own concentration in the cell. This form of negative regulation is called feedback inhibition.
A common type of control occurs when a molecule other than a substrate specifically binds to an enzyme at a special regulatory site, altering the rate at which the enzyme converts its substrate to product. In feedback inhibition, for example, an enzyme acting early in a reaction pathway is inhibited by a molecule produced later in that pathway. Thus, whenever large quantities of the final product begin to accumulate, the product binds to an earlier enzyme and slows down its catalytic action, limiting further entry of substrates into that reaction pathway (Figure 4−42). Where pathways branch or intersect, there are usually multiple points of control by different final products, each of which regulates its own synthesis (Figure 4−43). Feedback inhibition can work almost instantaneously and is rapidly reversed when product levels fall.
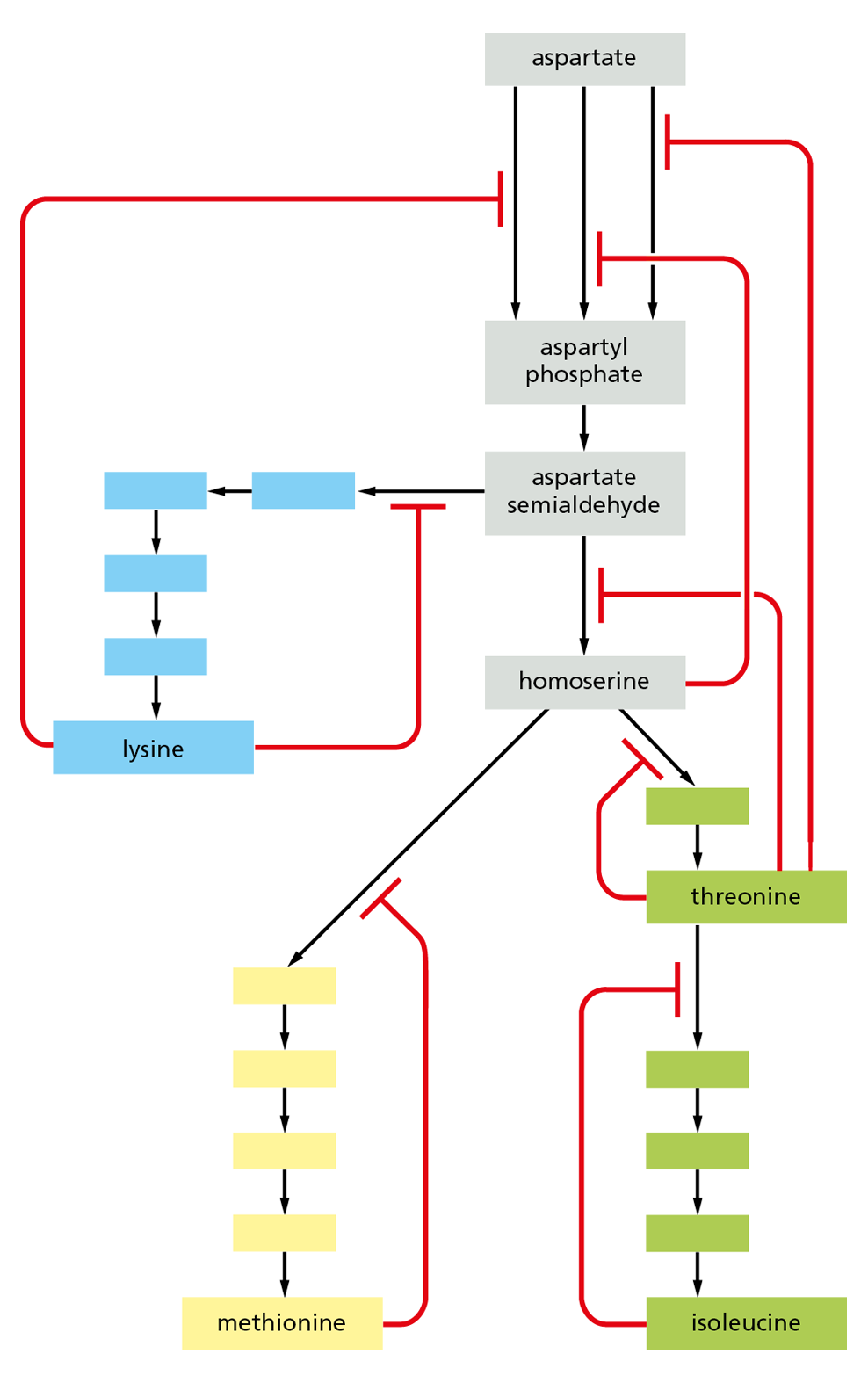
Figure 4−43 Feedback inhibition at multiple points regulates connected metabolic pathways. The biosynthetic pathways for four different amino acids in bacteria are shown, starting from the amino acid aspartate. The red lines indicate points at which products feed back to inhibit enzymes and the blank boxes represent intermediates in each pathway. In this example, each amino acid controls the first enzyme specific to its own synthesis, thereby limiting its own concentration and avoiding a wasteful buildup of intermediates. Some of the products also separately inhibit the initial set of reactions common to all the syntheses. Three different enzymes catalyze the initial reaction from aspartate to aspartyl phosphate, and each of these enzymes is inhibited by a different product.
Question 4–6
Consider the drawing in Figure 4−42. What will happen if, instead of the indicated feedback,
A. feedback inhibition from Z affects the step B → C only?
B. feedback inhibition from Z affects the step Y → Z only?
C. Z is a positive regulator of the step B → X?
D. Z is a positive regulator of the step B → C?
For each case, discuss how useful these regulatory schemes would be for a cell.
Feedback inhibition is a form of negative regulation: it prevents an enzyme from acting. Enzymes can also be subject to positive regulation, in which the enzyme’s activity is stimulated by a regulatory molecule rather than being suppressed. Positive regulation occurs when a product in one branch of the metabolic maze stimulates the activity of an enzyme in another pathway. But how do these regulatory molecules change an enzyme’s activity?
Allosteric Enzymes Have Two or More Binding Sites That Influence One Another
Feedback inhibition was initially puzzling to those who discovered it, in part because the regulatory molecule often has a shape that is totally different from the shape of the enzyme’s preferred substrate. Indeed, when this form of regulation was discovered in the 1960s, it was termed allostery (from the Greek allo, “other,” and stere, “solid” or “shape”). Given the numerous, specific, noncovalent interactions that allow enzymes to interact with their substrates within the active site, it seemed likely that these regulatory molecules were binding somewhere else on the surface of the protein. As more was learned about feedback inhibition, researchers realized that many enzymes must contain at least two different binding sites: an active site that recognizes the substrates and one or more sites that recognize regulatory molecules. These sites must somehow “communicate” to allow the catalytic events at the active site to be influenced by the binding of the regulatory molecule at a separate location.
The interaction between sites that are located in different regions on a protein molecule is now known to depend on a conformational change in the protein. The binding of a ligand to one of the sites causes a shift in the protein’s structure from one folded shape to a slightly different folded shape, and this alters the shape of a second binding site that can be far away. Many enzymes have two conformations that differ in activity, each of which can be stabilized by the binding of a different ligand. During feedback inhibition, for example, the binding of an inhibitor at a regulatory site on a protein causes the protein to spend more time in a conformation in which its active site—located elsewhere in the protein—becomes less accommodating to the substrate molecule (Figure 4−44).
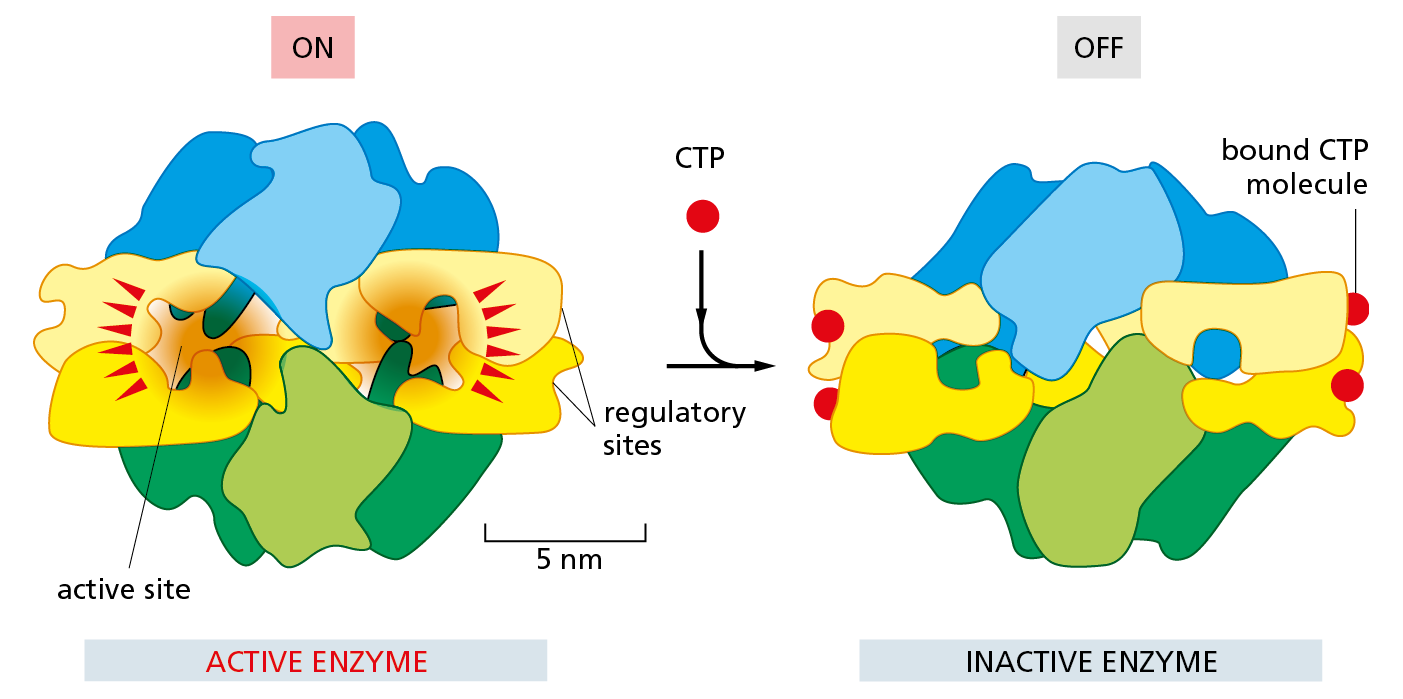
Figure 4−44 Feedback inhibition triggers a conformational change in an enzyme. Aspartate transcarbamoylase from E. coli, a large multisubunit enzyme used in early studies of allosteric regulation, catalyzes an important reaction that begins the synthesis of the pyrimidine ring of C, U, and T nucleotides (see Panel 2–7, pp. 78–79). One of the final products of this pathway, cytidine triphosphate (CTP), binds to the enzyme to turn it off whenever CTP is plentiful. This diagram shows the conformational change that occurs when the enzyme is turned off by CTP binding to its four regulatory sites, which are distinct from the active site where the substrate binds. Figure 4−10 shows the structure of aspartate transcarbamoylase as seen from the top. This figure depicts the enzyme as seen from the side.
As schematically illustrated in Figure 4–45A, many—if not most—protein molecules are allosteric: they can adopt two or more slightly different conformations, and their activity can be regulated by a shift from one to another. This is true not only for enzymes, but also for many other proteins as well. The chemistry involved here is extremely simple in concept. Because each protein conformation will have somewhat different contours on its surface, the protein’s binding sites for ligands will be altered when the protein changes shape. Each ligand will stabilize the conformation that it binds to most strongly. Therefore, at high enough concentrations, a ligand will tend to “switch” the population of proteins to the conformation that it favors (Figure 4−45B and C).
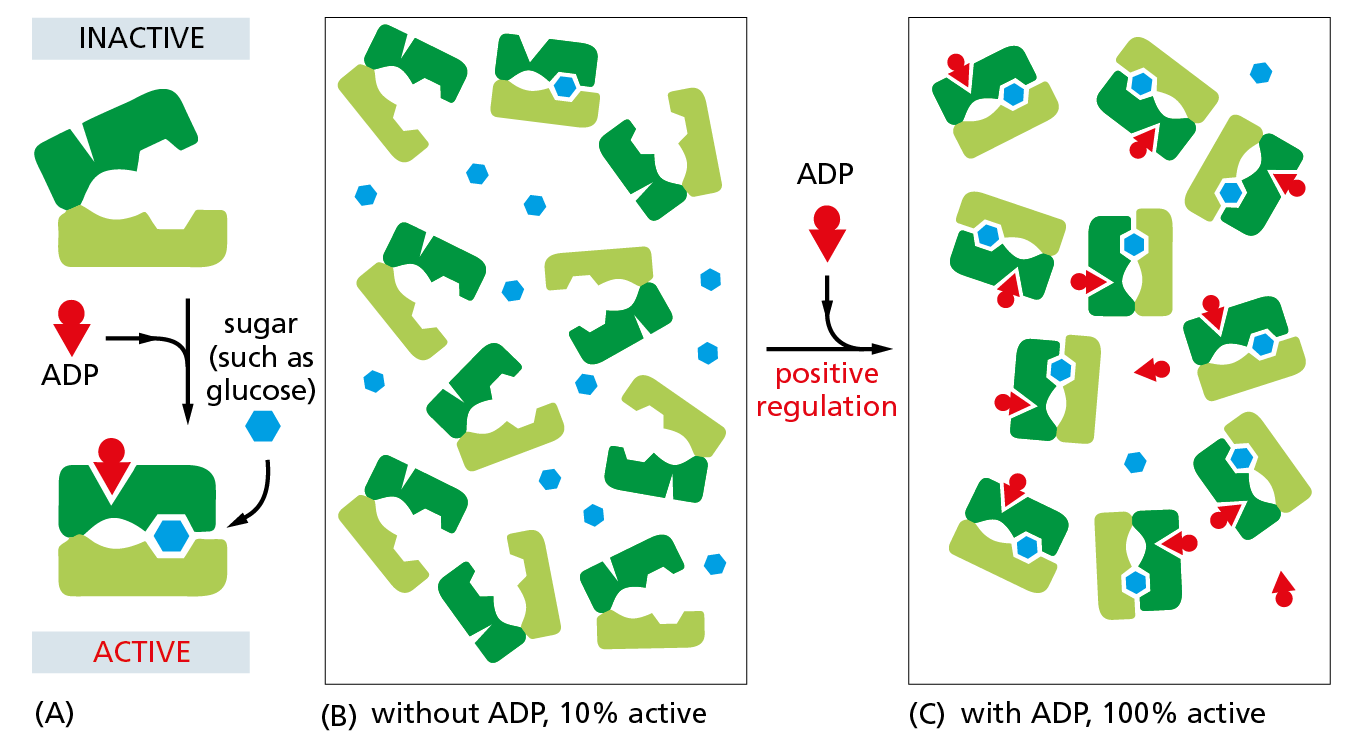
Figure 4−45 The binding of a regulatory ligand can change the equilibrium between two protein conformations. (A) Schematic diagram of a hypothetical, allosterically regulated enzyme for which a rise in the concentration of ADP molecules (red wedges) increases the rate at which the enzyme catalyzes the oxidation of sugar molecules (blue hexagons). (B) Due to thermal motions, the enzyme will spontaneously interconvert between the open (inactive) and closed (active) conformations shown in (A). But when ADP is absent, only a small fraction of the enzyme molecules will be present in the active conformation at any given time. As illustrated, most remain in the inactive conformation. (C) Because ADP can bind to the protein only in its closed, active conformation, an increase in ADP concentration locks nearly all of the enzyme molecules in the active form—an example of positive regulation. In cells, rising concentrations of ADP signal a depletion of ATP reserves; increased oxidation of sugars—in the presence of ADP—thus provides more energy for the synthesis of ATP from ADP.
Phosphorylation Can Control Protein Activity by Causing a Conformational Change
Another method that eukaryotic cells use to regulate protein activity involves attaching a phosphate group covalently to one or more of the protein’s amino acid side chains. Because each phosphate group carries two negative charges, the enzyme-catalyzed addition of a phosphate group can cause a conformational change by, for example, attracting a cluster of positively charged amino acid side chains from somewhere else in the same protein. This structural shift can, in turn, affect the binding of ligands elsewhere on the protein surface, thereby altering the protein’s activity. Removal of the phosphate group by a second enzyme will return the protein to its original conformation and restore its initial activity.
Reversible protein phosphorylation controls the activity of many types of proteins in eukaryotic cells. This form of regulation is used so extensively that more than one-third of the 10,000 or so proteins in a typical mammalian cell are phosphorylated at any one time. The addition and removal of phosphate groups from specific proteins often occur in response to signals that specify some change in a cell’s state. For example, the complicated series of events that takes place as a eukaryotic cell divides is timed largely in this way (discussed in Chapter 18). And many of the intracellular signaling pathways activated by extracellular signals depend on a network of protein phosphorylation events (discussed in Chapter 16).
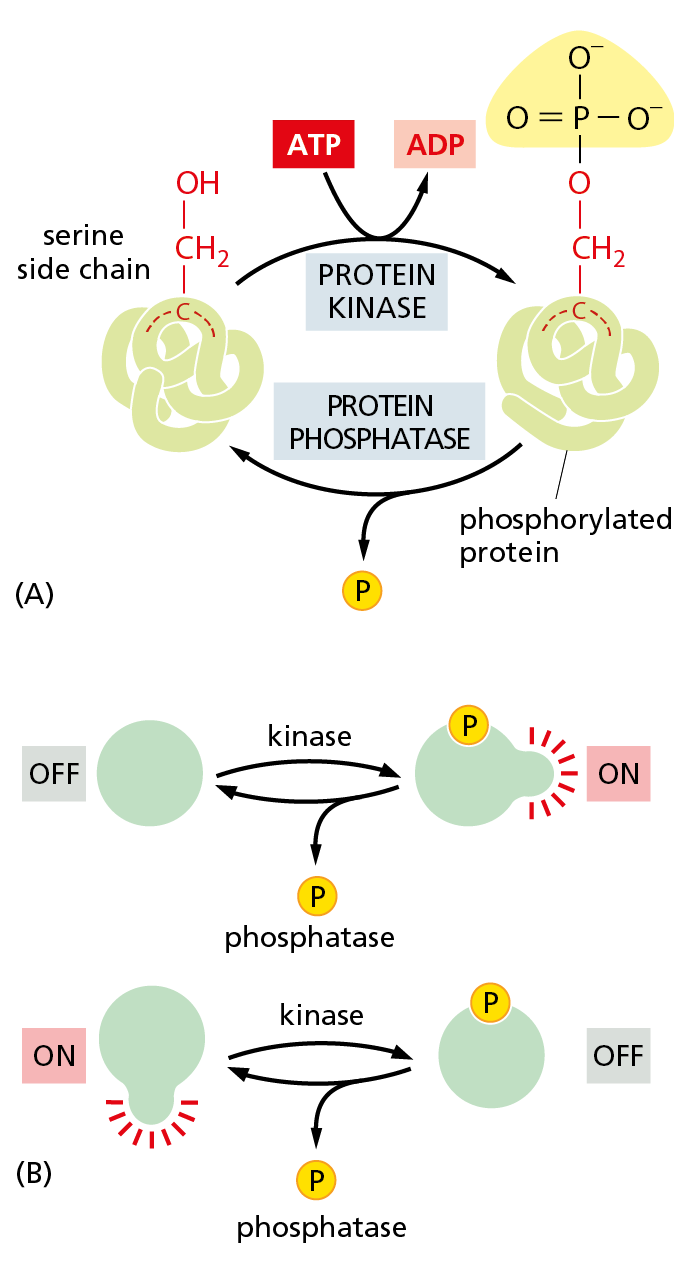
Figure 4−46 Protein phosphorylation is a very common mechanism for regulating protein activity. Many thousands of proteins in a typical eukaryotic cell are modified by the covalent addition of one or more phosphate groups. (A) The general reaction, shown here, entails transfer of a phosphate group from ATP to an amino acid side chain of the target protein by a protein kinase. Removal of the phosphate group is catalyzed by a second enzyme, a protein phosphatase. In this example, the phosphate is added to a serine side chain; in other cases, the phosphate is instead linked to the –OH group of a threonine or tyrosine side chain. (B) Phosphorylation can either increase or decrease the protein’s activity, depending on the site of phosphorylation and the structure of the protein.
Protein phosphorylation involves the enzyme-catalyzed transfer of the terminal phosphate group of ATP to the hydroxyl group on a serine, threonine, or tyrosine side chain of the protein. This reaction is catalyzed by a protein kinase. The reverse reaction—removal of the phosphate group, or dephosphorylation—is catalyzed by a protein phosphatase (Figure 4−46A). Phosphorylation can either stimulate protein activity or inhibit it, depending on the protein involved and the site of phosphorylation (Figure 4−46B). Cells contain hundreds of different protein kinases, each responsible for phosphorylating a different protein or set of proteins. Cells also contain a smaller set of different protein phosphatases; some of these are highly specific and remove phosphate groups from only one or a few proteins, whereas others act on a broad range of proteins. The state of phosphorylation of a protein at any moment in time, and thus its activity, will depend on the relative activities of the protein kinases and phosphatases that act on it.
Phosphorylation can take place in a continuous cycle, in which a phosphate group is rapidly added to—and rapidly removed from—a particular side chain. Such phosphorylation cycles allow proteins to switch quickly from one state to another. The more swiftly the cycle is “turning,” the faster the concentration of a phosphorylated protein can change in response to a sudden stimulus. Although keeping the cycle turning costs energy—because ATP is hydrolyzed with each phosphorylation—many enzymes in the cell undergo this speedy, cyclic form of regulation.
Covalent Modifications Also Control the Location and Interaction of Proteins
Phosphorylation can do more than control a protein’s activity; it can create docking sites where other proteins can bind, thus promoting the assembly of proteins into larger complexes. For example, when extracellular signals stimulate a class of cell-surface, transmembrane proteins called receptor tyrosine kinases, they cause the receptor proteins to phosphorylate themselves on certain tyrosines. The phosphorylated tyrosines then serve as docking sites for the binding and activation of a set of intracellular signaling proteins, which transmits the message to the cell interior and changes the behavior of the cell (see Figure 16−29).
Phosphorylation is not the only form of covalent modification that can affect a protein’s function. Many proteins are modified by the addition of an acetyl group to a lysine side chain, including the histones discussed in Chapter 5. And the addition of the fatty acid palmitate to a cysteine side chain drives a protein to associate with cell membranes. Attachment of ubiquitin, a 76-amino-acid polypeptide, can target a protein for degradation, as we discuss in Chapter 7. More than 100 types of covalent modifications can occur in the cell, each playing its own role in regulating protein function. Each of these modifying groups is enzymatically added or removed depending on the needs of the cell.
A large number of proteins are modified on more than one amino acid side chain. The p53 protein, which plays a central part in controlling how a cell responds to DNA damage and other stresses, can be covalently modified at 20 sites (Figure 4−47). Because an enormous number of combinations of these 20 modifications is possible, the protein’s behavior can in principle be altered in a huge number of ways.
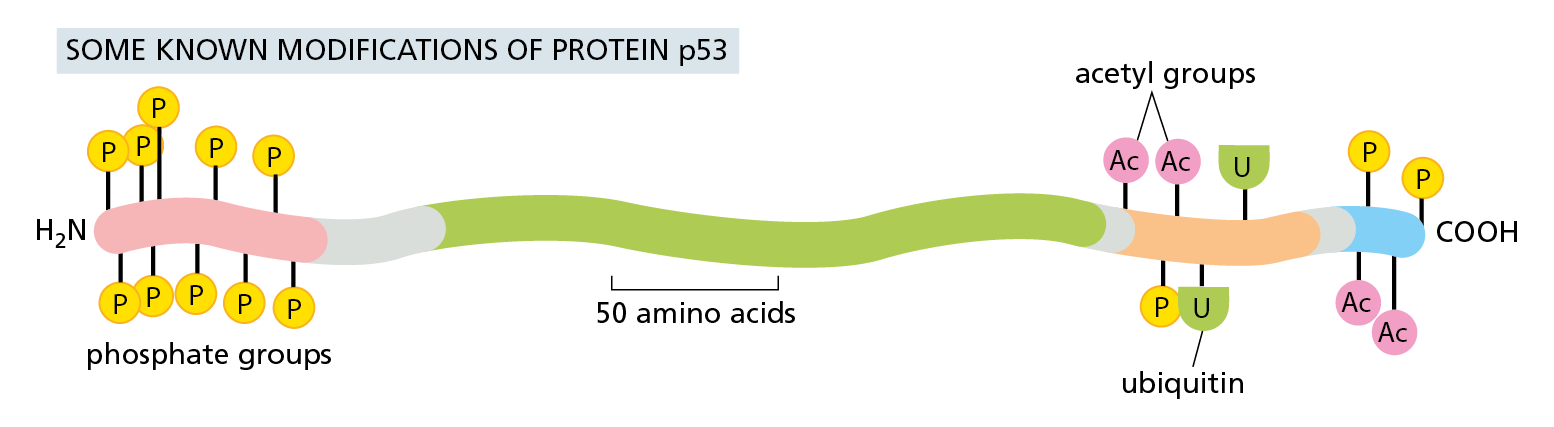
Figure 4−47 The modification of a protein at multiple sites can control the protein’s behavior. This diagram shows some of the covalent modifications that control the activity and degradation of p53, a protein of nearly 400 amino acids. p53 is an important transcription regulator that regulates a cell’s response to damage (discussed in Chapter 18). Not all of these modifications will be present at the same time. Colors along the body of the protein represent distinct protein domains, including one that binds to DNA (green) and one that activates gene transcription (pink). All of the modifications shown are located within relatively unstructured regions of the polypeptide chain.
The set of covalent modifications that a protein contains at any moment constitutes an important form of regulation. The attachment or removal of these modifying groups can change a protein’s activity or stability, its binding partners, or its location inside the cell. Covalent modifications thus enable the cell to make optimal use of the proteins it produces, and they allow the cell to respond rapidly to changes in its environment.
Regulatory GTP-Binding Proteins Are Switched On and Off by the Gain and Loss of a Phosphate Group
Eukaryotic cells have a second way to regulate protein activity by phosphate addition and removal. In this case, however, the phosphate is not enzymatically transferred from ATP to the protein. Instead, the phosphate is part of a guanine nucleotide—guanosine triphosphate (GTP)—that binds tightly various types of GTP-binding proteins. These proteins act as molecular switches: they are in their active conformation when GTP is bound, but they can hydrolyze this GTP to GDP—which releases a phosphate and flips the protein to an inactive conformation (Movie 4.10). As with protein phosphorylation, this process is reversible: the active conformation is regained by dissociation of the GDP, followed by the binding of a fresh molecule of GTP (Figure 4−48).
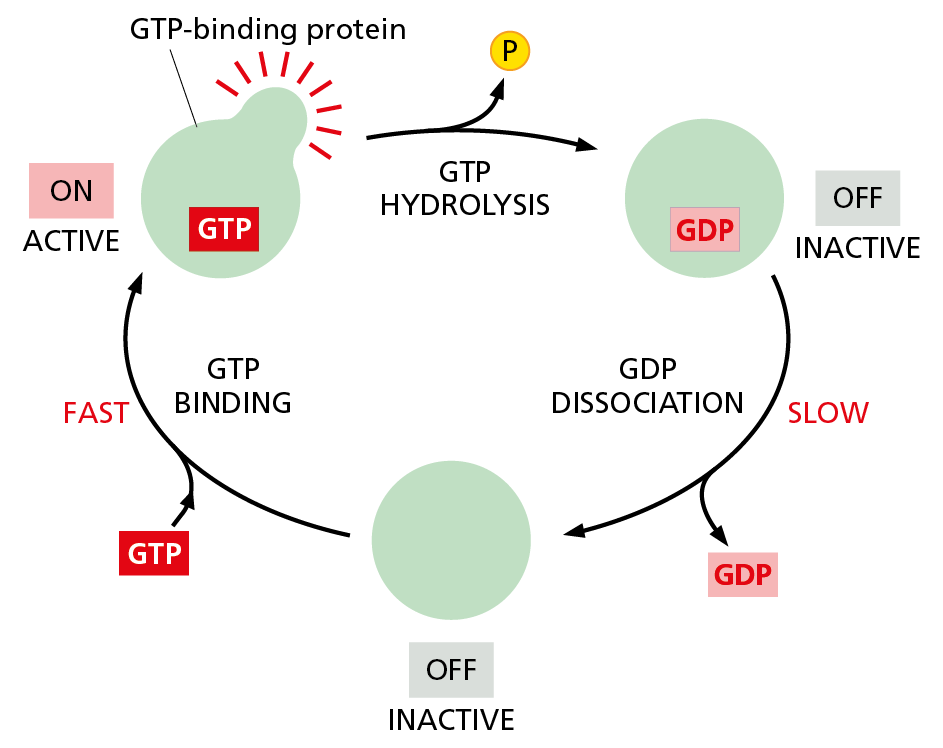
Figure 4−48 Many different GTP-binding proteins function as molecular switches. A GTP-binding protein requires the presence of a tightly bound GTP molecule to be active. The active protein can shut itself off by hydrolyzing its bound GTP to GDP and inorganic phosphate (Pi), which converts the protein to an inactive conformation. To reactivate the protein, the tightly bound GDP must dissociate. As explained in Chapter 16, this dissociation is a slow step that can be greatly accelerated by important regulatory proteins called guanine nucleotide exchange factors (GEFs). As indicated, once the GDP dissociates, a molecule of GTP quickly replaces it, returning the protein to its active conformation.
Question 4–7
Either protein phosphorylation or the binding of a nucleotide (such as ATP or GTP) can be used to regulate a protein’s activity. What do you suppose are the advantages of each form of regulation?
Hundreds of GTP-binding proteins function as molecular switches in cells. The dissociation of GDP and its replacement by GTP, which turns the switch on, is often stimulated in response to cell signals. The GTP-binding proteins activated in this way in turn bind to other proteins to regulate their activities. The crucial role GTP-binding proteins play in intracellular signaling pathways is discussed in detail in Chapter 16.
ATP Hydrolysis Allows Motor Proteins to Produce Directed Movements in Cells
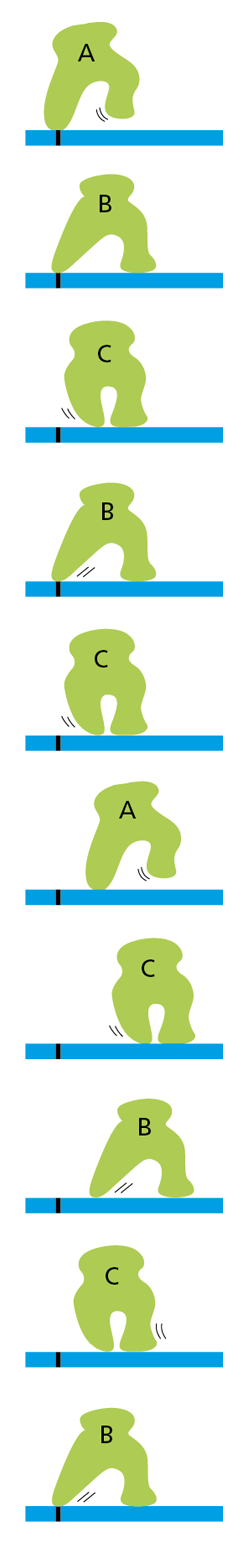
Figure 4−49 Changes in conformation can allow a protein to “walk” along a cytoskeletal filament. This protein cycles between three different conformations (A, B, and C) as it moves along the filament. But, without an input of energy to drive its movement in a single direction, the protein can only wander randomly back and forth, ultimately getting nowhere.
We have seen how conformational changes in proteins play a central part in enzyme regulation and cell signaling. But conformational changes also play another important role in the operation of the eukaryotic cell: they enable certain specialized proteins to drive directed movements of cells and their components. These motor proteins generate the forces responsible for muscle contraction and most other eukaryotic cell movements. They also power the intracellular movements of organelles and macromolecules. For example, they help move chromosomes to opposite ends of the cell during mitosis (discussed in Chapter 18), and they move organelles along cytoskeletal tracks (discussed in Chapter 17).
But how can the changes in shape experienced by proteins be used to generate such orderly movements? A protein that is required to walk along a cytoskeletal fiber, for example, can move by undergoing a series of conformational changes. However, with nothing to drive these changes in one direction or the other, the shape changes will be reversible and the protein will wander randomly back and forth (Figure 4−49).
To force the protein to proceed in a single direction, the conformational changes must be unidirectional. To achieve such directionality, one of the steps must be made irreversible. For most proteins that are able to move in a single direction for long distances, this irreversibility is achieved by coupling one of the conformational changes to the hydrolysis of an ATP molecule that is tightly bound to the protein—which is why motor proteins are also ATPases. A great deal of free energy is released when ATP is hydrolyzed, making it very unlikely that the protein will undergo a reverse shape change—as required for moving backward. (Such a reversal would require that the ATP hydrolysis be reversed, by adding a phosphate molecule to ADP to form ATP.) As a consequence, the protein moves steadily forward (Figure 4−50).
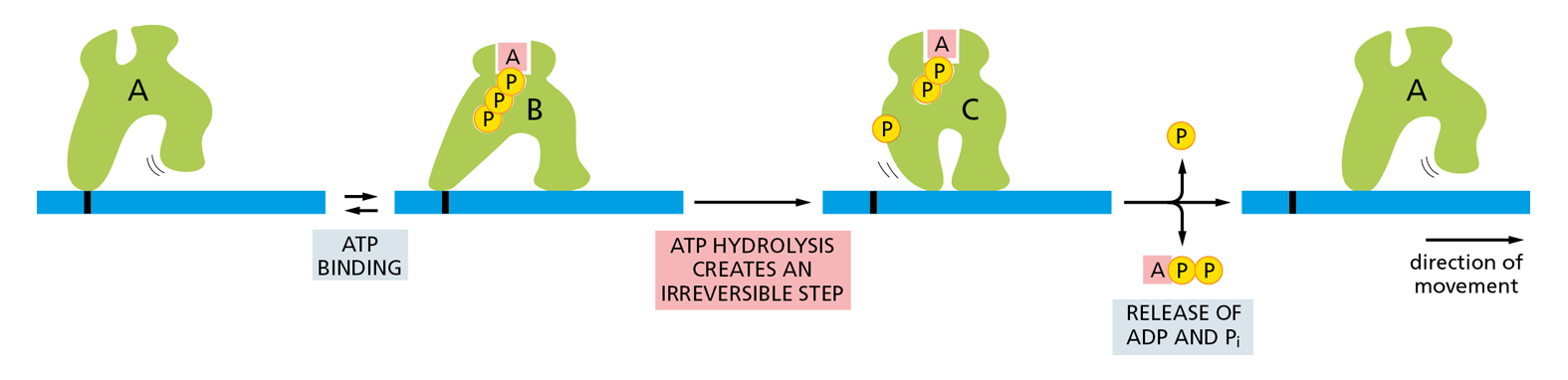
Figure 4−50 A schematic model of how a motor protein uses ATP hydrolysis to move in one direction along a cytoskeletal filament. An orderly transition among three conformations is driven by the hydrolysis of a bound ATP molecule and the release of the products, ADP and inorganic phosphate (Pi). Because these transitions are coupled to the hydrolysis of ATP, the entire cycle is essentially irreversible. Through repeated cycles, the protein moves continuously to the right along the filament.
Many different motor proteins generate directional movement by using the hydrolysis of a tightly bound ATP molecule to drive an orderly series of conformational changes. These movements can be rapid: the muscle motor protein myosin walks along actin filaments at about 6 μm/sec during muscle contraction (discussed in Chapter 17).
Proteins Often Form Large Complexes That Function as Machines
As proteins progress from being small, with a single domain, to being larger with multiple domains, the functions they can perform become more elaborate. The most complex tasks are carried out by large protein assemblies formed from many protein molecules. Now that it is possible to reconstruct biological processes in cell-free systems in a test tube, it is clear that each central process in a cell—including DNA replication, gene transcription, protein synthesis, vesicle budding, and transmembrane signaling—is catalyzed by a highly coordinated, linked set of many proteins. For most such protein machines, the hydrolysis of bound nucleoside triphosphates (ATP or GTP) drives an ordered series of conformational changes in some of the individual protein subunits, enabling the ensemble of proteins to move coordinately (Figure 4−51). In these machine-like complexes, the appropriate enzymes can be positioned to carry out successive reactions in a series—as during the synthesis of proteins on a ribosome, for example (discussed in Chapter 7). And during cell division, a large protein machine moves rapidly along DNA to replicate the DNA double helix (discussed in Chapter 6 and shown in Movie 6.3 and Movie 6.4).
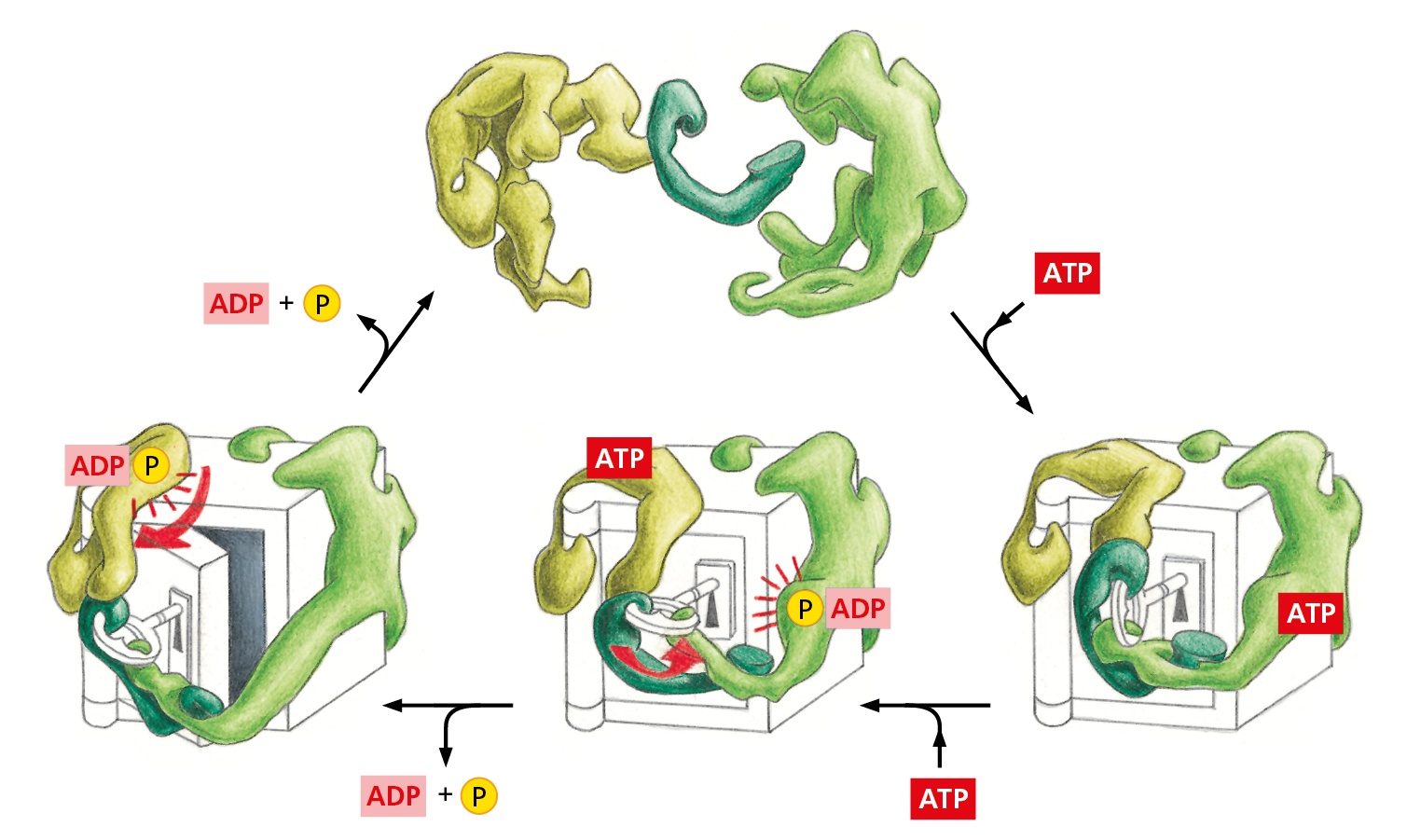
Figure 4−51 “Protein machines” can carry out complex functions. These machines are made of individual proteins that collaborate to perform a specific task (Movie 4.11). The movement of proteins is often coordinated and made unidirectional by the hydrolysis of a bound nucleotide such as ATP. Conformational changes of this type are especially useful to the cell if they occur in a large protein assembly in which the activities of several different protein molecules can be coordinated by the movements within the complex, as schematically illustrated here.
Question 4–8
Explain why the hypothetical enzymes in Figure 4−51 have a great advantage in opening the safe if they work together in a protein complex, as opposed to working individually in an unlinked, sequential manner.
A large number of different protein machines have evolved to perform many critical biological tasks. Cells make wide use of protein machines for the same reason that humans have invented mechanical and electronic machines: for almost any job, manipulations that are spatially and temporally coordinated through linked processes are much more efficient than is the sequential use of individual tools.
Many Interacting Proteins Are Brought Together by Scaffolds
We have seen that proteins rely on interactions with other molecules to carry out their biological functions. Enzymes bind substrates and regulatory ligands—many of which are generated by other enzymes in the same reaction pathway. Receptor proteins in the plasma membrane, when activated by extracellular ligands, can recruit a set of intracellular signaling proteins that interact with and activate one another, propagating the signal to the cell interior. In addition, the proteins involved in DNA replication, gene transcription, DNA repair, and protein synthesis form protein machines that carry out these complex and crucial tasks with great efficiency.
But how do proteins find the appropriate partners—and the sites where they are needed—within the crowded conditions inside the cell (see Figure 3−22)? Many protein complexes are brought together by scaffold proteins, large molecules that contain binding sites recognized by multiple proteins. By binding a specific set of interacting proteins, a scaffold can greatly enhance the rate of a particular chemical reaction or cell process, while also confining this chemistry to a particular area of the cell—for example, drawing signaling proteins to the plasma membrane.
Although some scaffolds are rigid, the most abundant scaffolds in cells are very elastic. Because they contain long unstructured regions that allow them to bend and sway, these scaffolds serve as flexible tethers that greatly enhance the collisions between the proteins that are bound to them (Figure 4−52). Some other scaffolds are not proteins but long molecules of RNA. We encounter these RNA scaffolds when we discuss RNA synthesis and processing in Chapter 7.
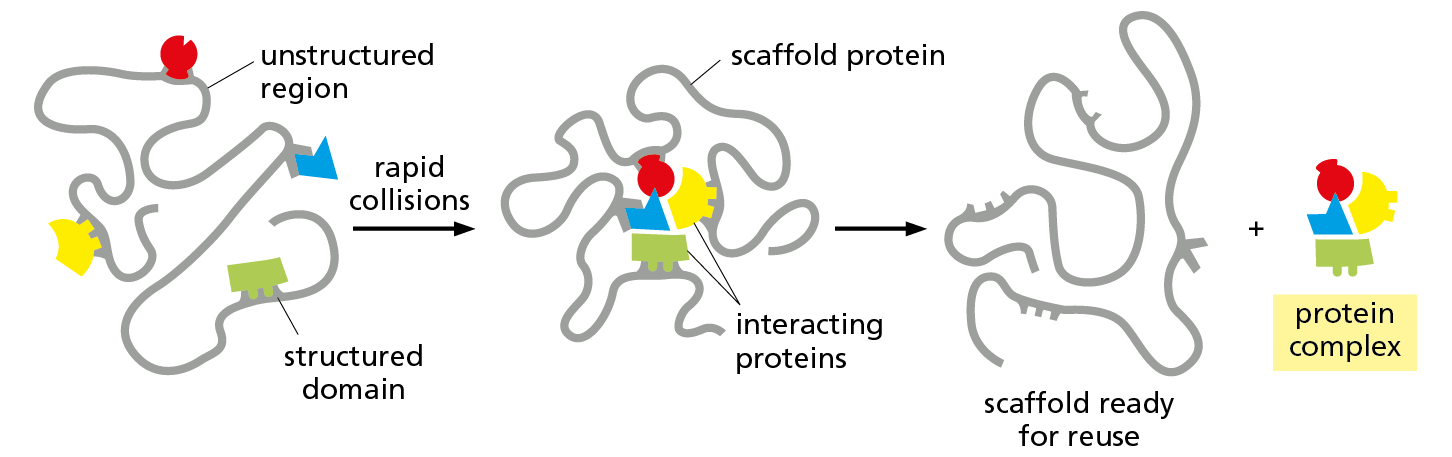
Figure 4−52 Scaffold proteins can concentrate interacting proteins in the cell. In this hypothetical example, each of a set of interacting proteins is bound to a specific structured domain within a long, otherwise unstructured scaffold protein. The unstructured regions of the scaffold act as flexible tethers, and they enhance the rate of formation of the functional complex by promoting the rapid, random collision of the proteins bound to the scaffold.
Scaffolds allow proteins to be assembled and activated only when and where they are needed. Nerve cells, for example, deploy large, flexible scaffold proteins—some more than 1000 amino acids in length—to organize the specialized proteins involved in transmitting and receiving the signals that carry information from one nerve cell to the next. These proteins cluster beneath the plasma membranes of communicating nerve cells (see Figure 4–54), allowing them both to transmit and to respond to the appropriate messages when stimulated to do so.
Weak Interactions Between Macromolecules Can Produce Large Biochemical Subcompartments in Cells
The aggregates formed by sets of proteins, RNAs, and protein machines can grow quite large, producing distinct biochemical compartments within the cell. The largest of these is the nucleolus—the nuclear compartment in which ribosomal RNAs are transcribed and ribosomal subunits are assembled. This cell structure, which is formed when the chromosomes that carry the ribosomal genes come together during interphase (see Figure 5−17), is large enough to be seen in a light microscope. Smaller, transient structures assemble as needed in the nucleus to generate “factories” that carry out DNA replication, DNA repair, or mRNA production (see Figure 7–24). In addition, specific mRNAs are sequestered in cytoplasmic granules that help to control their use in protein synthesis.
The general term used to describe such assemblies, many of which contain both protein and RNA, is an intracellular condensate. Some of these condensates, including the nucleolus, can take the form of spherical, liquid droplets that can be seen to break up and fuse (Figure 4–53). Although these condensates resemble the sort of phase-separated compartments that form when oil and water mix, their interior makeup is complex and structured. Some are based on amyloid structures, reversible assemblies of stacked β sheets that come together to produce a “hydrogel” that pulls other molecules into the condensate (Figure 4−54). Amyloid-forming proteins thus have functional roles in cells. But for a handful of these amyloid-forming proteins, mutation or perturbation can lead to neurological disease, which is how some of them were initially discovered.
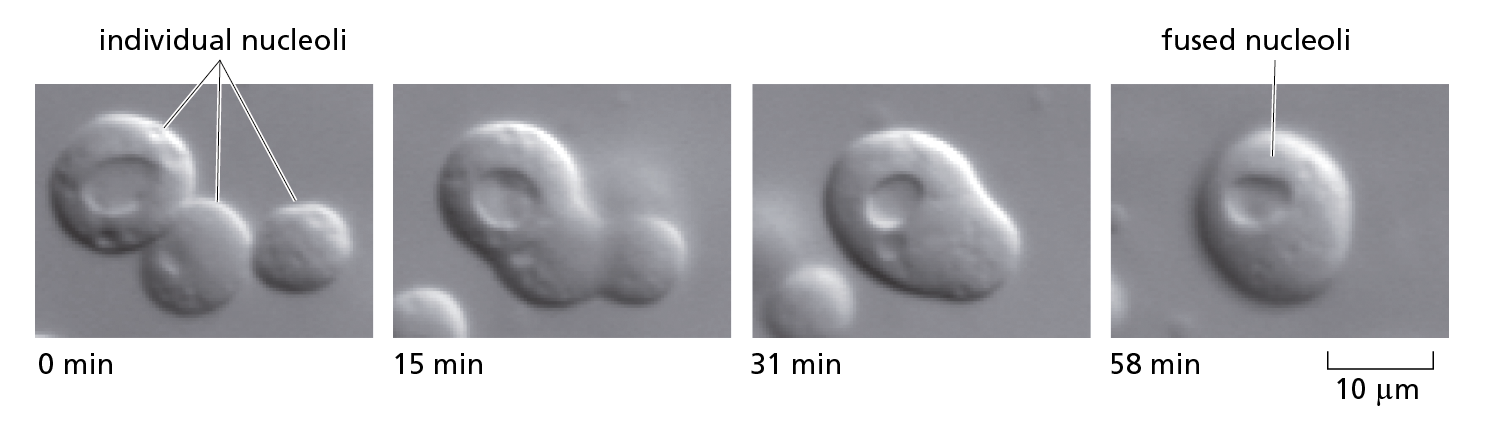
Figure 4−53 Spherical, liquid-drop-like nucleoli can be seen to fuse in the light microscope. In these experiments, the nucleoli are present inside a nucleus that has been dissected from Xenopus oocytes and placed under oil on a microscope slide. Here, three nucleoli are seen fusing to form one larger nucleolus (Movie 4.12). A very similar process occurs following each round of division, when small nucleoli initially form on multiple chromosomes, but then coalesce to form a single, large nucleolus. (From C.P. Brangwynne, T.J. Mitchison, and A.A. Hyman, Proc. Natl. Acad. Sci. USA 108:4334–4339, 2011.)
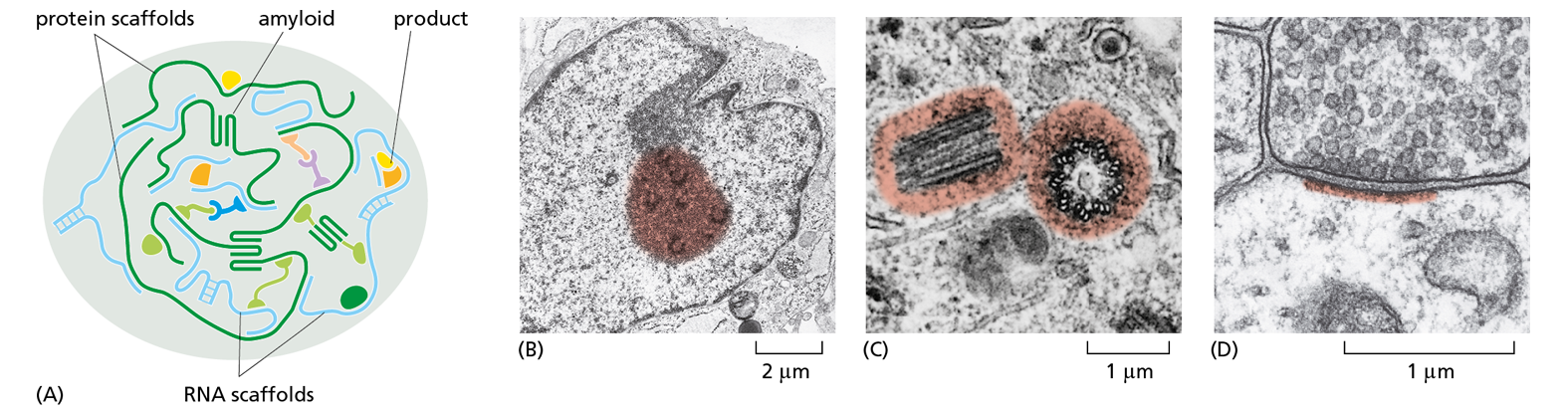
Figure 4−54 Intracellular condensates can form biochemical subcompartments in cells. These large aggregates form as a result of multiple weak binding interactions between scaffolds and other macromolecules. When these macromolecule–macromolecule interactions become sufficiently strong, a “phase separation” occurs. This creates two distinct aqueous compartments, in one of which the interacting molecules are densely aggregated. Such intracellular condensates concentrate a select set of macromolecules, thereby producing regions with a special biochemistry without the use of an encapsulating membrane.
(A) Schematic illustration of a phase-separated intracellular condensate. These condensates can create a factory that catalyzes the formation of a specific type of product, or they can serve to store important entities, such as specific mRNA molecules, for later use. As shown, reversible amyloid structures often help to create these aggregates. These β-sheet structures form between regions of unstructured amino acid sequence within the larger protein scaffolds.
(B–D) Three examples that illustrate how intracellular condensates (colorized regions) are thought to be used by cells. (B) Inside the interphase nucleus, the nucleolus is a large factory that produces ribosomes. In addition, many scattered RNA production factories concentrate the protein machines that transcribe the genome. (C) In the cytoplasm, a matrix forms the centrosome that nucleates the assembly of microtubules. (D) In a patch underlying the plasma membrane at the synapse where communicating nerve cells touch, multiple interacting scaffolds produce large protein assemblies; these create a local biochemistry that makes possible memory formation and storage in the nerve cell network. (B, courtesy of E.G. Jordan and J. McGovern; C, from M. McGill, D.P. Highfield, T.M. Monahan, and B.R. Brinkley, J. Ultrastruct. Res. 57:43–53, 1976. With permission from Elsevier; D, courtesy of Cedric Raine.)
- feedback inhibition
A form of metabolic control in which the end product of a chain of enzymatic reactions reduces the activity of an enzyme early in the pathway.
- allosteric
Describes a protein that can exist in multiple conformations depending on the binding of a molecule (ligand) at a site other than the catalytic site; such changes from one conformation to another often alter the protein’s activity or ligand affinity.
- protein phosphorylation
The covalent addition of a phosphate group to a side chain of a protein, catalyzed by a protein kinase; serves as a form of regulation that usually alters the activity or properties of the target protein.
- protein kinase
Enzyme that catalyzes the transfer of a phosphate group from ATP to a specific amino acid side chain on a target protein.
- protein phosphatase
Enzyme that catalyzes the removal of a phosphate group from a protein, often with high specificity for the phosphorylated site.
- GTP-binding protein
Intracellular signaling protein whose activity is determined by its association with either GTP or GDP. Includes both trimeric G proteins and monomeric GTPases, such as Ras.
- motor protein
Protein such as myosin or kinesin that uses energy derived from the hydrolysis of a tightly bound ATP molecule to propel itself along a protein filament or polymeric molecule.
- protein machine
Assembly of protein molecules that operates as a cooperative unit to perform a complex series of biological activities, such as replicating DNA.
- scaffold protein
Protein with multiple binding sites for other macromolecules, holding them in a way that speeds up their functional interactions.
- intracellular condensate
A large aggregate of phase-separated macromolecules that creates a region with a special biochemistry without the use of an encapsulating membrane.