DNA REPAIR
Maintaining the genetic stability that an organism needs for its survival requires not only an extremely accurate mechanism for replicating DNA but also mechanisms for repairing the many accidental lesions that DNA continually suffers. Most such spontaneous changes in DNA are temporary because they are immediately corrected by a set of processes that are collectively called DNA repair. Of the tens of thousands of random changes created every day in the DNA of a human cell by heat, metabolic accidents, radiation of various sorts, and exposure to substances in the environment, only a few (less than 0.02%) accumulate as permanent mutations in the DNA sequence. The rest are eliminated with remarkable efficiency by DNA repair.
The importance of DNA repair is evident from the large investment that cells make in the enzymes that carry it out: several percent of the coding capacity of most genomes is devoted solely to DNA repair functions. The importance of DNA repair is also demonstrated by the increased rate of mutation that follows the inactivation of a DNA repair gene. Many DNA repair proteins and the genes that encode them—which we now know operate in a wide range of organisms, including humans—were originally identified in bacteria by the isolation and characterization of mutants that displayed an increased mutation rate or an increased sensitivity to DNA-damaging agents.
Studies of the consequences of a diminished capacity for DNA repair in humans have linked many human diseases with decreased repair (Table 5–2). Thus, we saw previously that defects in a human gene whose product normally functions to repair the mismatched base pairs resulting from DNA replication errors can lead to an inherited predisposition to cancers of the colon and some other organs, caused by an increased mutation rate. In another human disease, xeroderma pigmentosum (XP), the afflicted individuals have an extreme sensitivity to ultraviolet radiation because they are unable to repair the damage to DNA caused by this component of sunlight. This repair defect results in an increased mutation rate that leads to serious skin lesions and a greatly increased susceptibility to skin cancers. Finally, mutations in the Brca1 and Brca2 genes compromise a type of DNA repair known as homologous recombination and are a major cause of hereditary breast and ovarian cancers.
|
TABLE 5–2 Some Inherited Human Syndromes with Defects in DNA Repair |
||
|
Name of syndrome or responsible genes |
Phenotype |
Enzyme or process affected |
|
Msh2, Msh3, Msh6, Mlh1, Pms2 |
Colon cancer |
Mismatch repair |
|
Polymerase proofreading–associated polyposis |
Colon cancer |
Proofreading by DNA polymerase ε |
|
Aicardi–Goutières syndrome |
Encephalopathy, neurological dysfunction, genome instability |
Removal of misincorporated ribonucleotides in DNA |
|
Xeroderma pigmentosum (XP) groups A–G |
Skin cancer, UV sensitivity, neurological abnormalities |
Nucleotide excision repair |
|
Cockayne syndrome |
UV sensitivity, developmental abnormalities |
Coupling of nucleotide excision repair to transcription |
|
XP variant |
UV sensitivity, skin cancer |
Translesion synthesis by DNA polymerase η |
|
Ataxia telangiectasia (AT) |
Leukemia, lymphoma, γ-ray sensitivity, genome instability |
ATM protein, a protein kinase activated by double-strand DNA breaks |
|
Seckel syndrome |
Dwarfism, microcephaly |
ATR protein, a protein kinase activated by single-strand DNA breaks |
|
Brca1 |
Breast and ovarian cancer |
Repair by homologous recombination |
|
Brca2 |
Breast, ovarian, prostate, and pancreatic cancer |
Repair by homologous recombination |
|
Ataxia-telangiectasia-like disorder (ATLD) |
Leukemia, lymphoma, γ-ray sensitivity, genome instability |
Mre11 protein, required for processing double-strand DNA breaks |
|
Werner syndrome |
Premature aging, cancer at several sites, genome instability |
Accessory 3′-exonuclease and DNA helicase used in repair |
|
Bloom syndrome |
Cancer at several sites, stunted growth, genome instability |
DNA helicase needed for recombination |
|
Fanconi anemia groups A–W |
Congenital abnormalities, leukemia, genome instability |
DNA interstrand cross-link repair |
|
46BR patient |
Hypersensitivity to DNA-damaging agents, genome instability |
DNA ligase I |
Without DNA Repair, Spontaneous DNA Damage Would Rapidly Change DNA Sequences
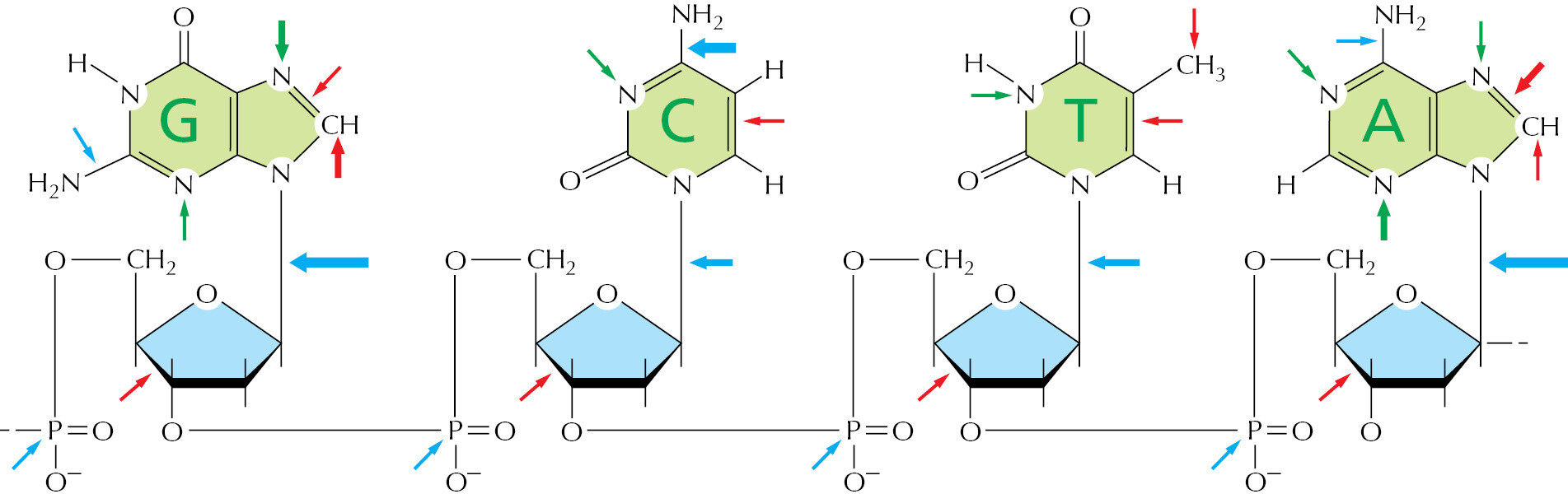
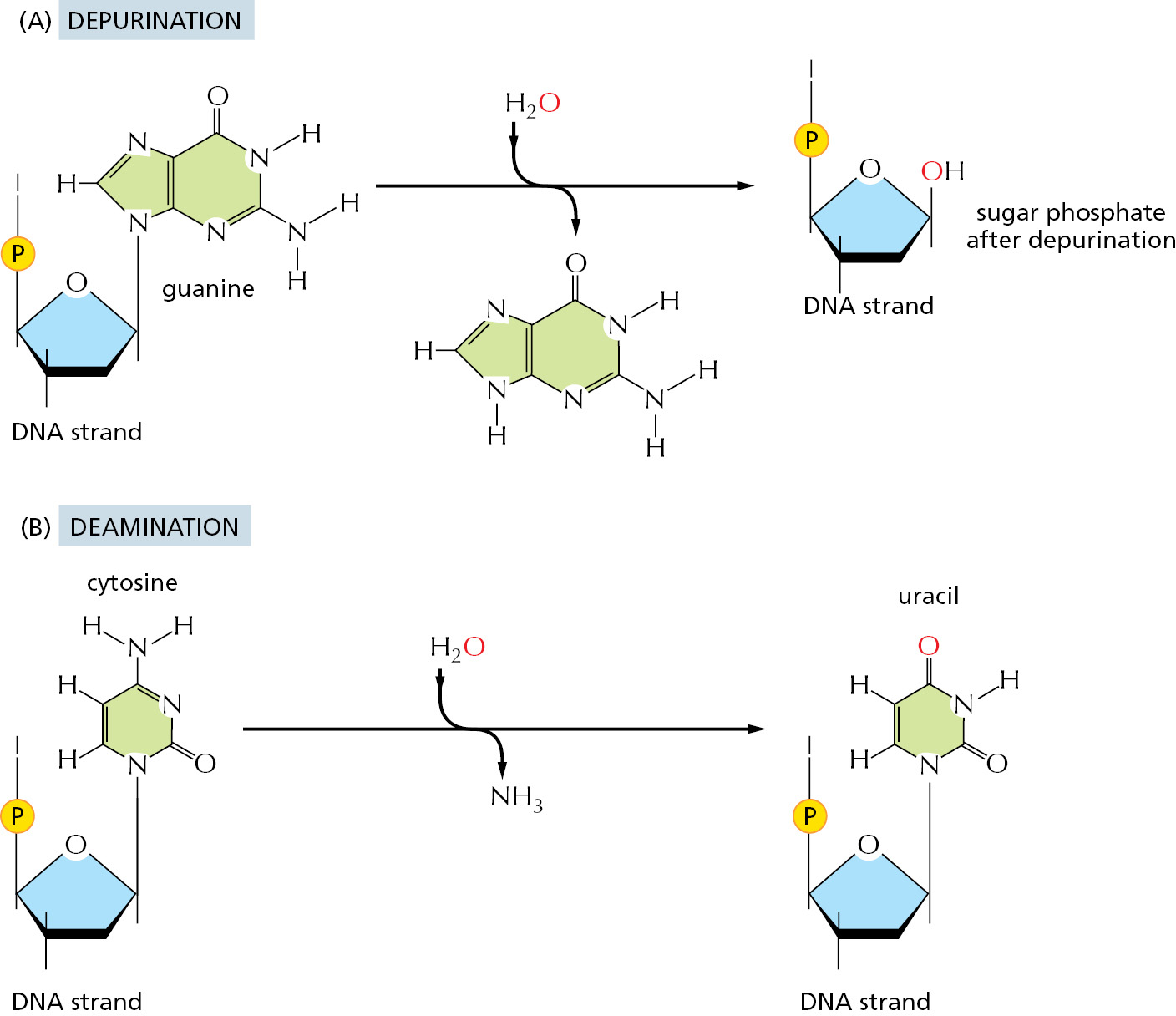
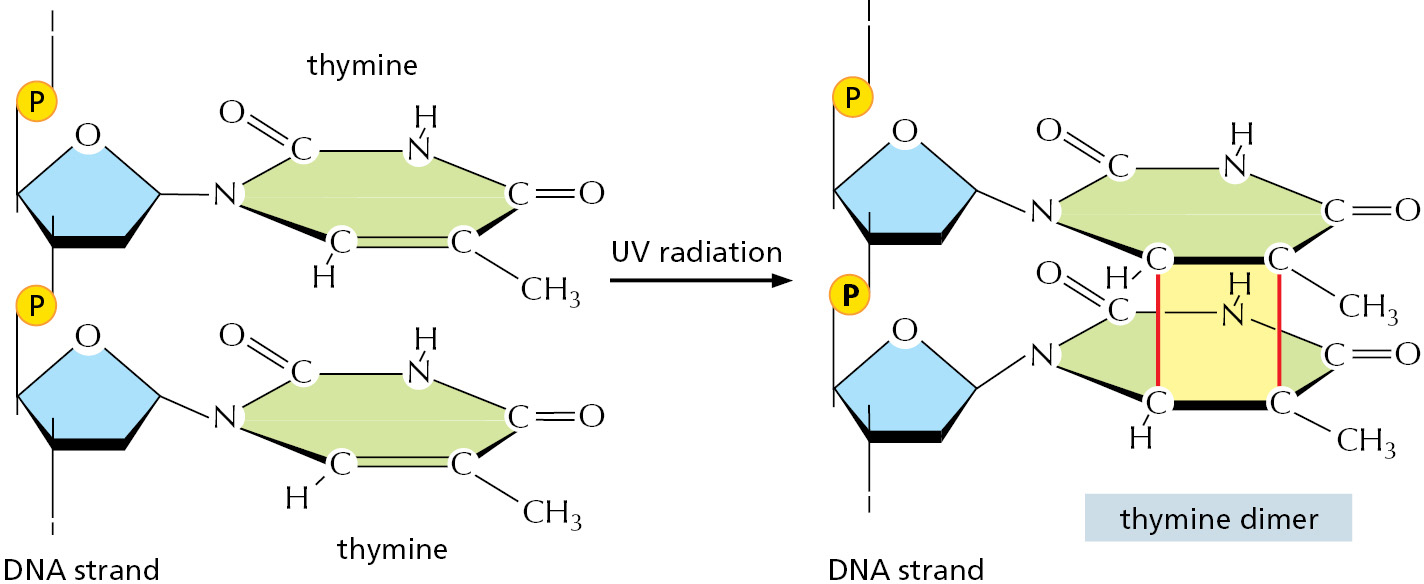
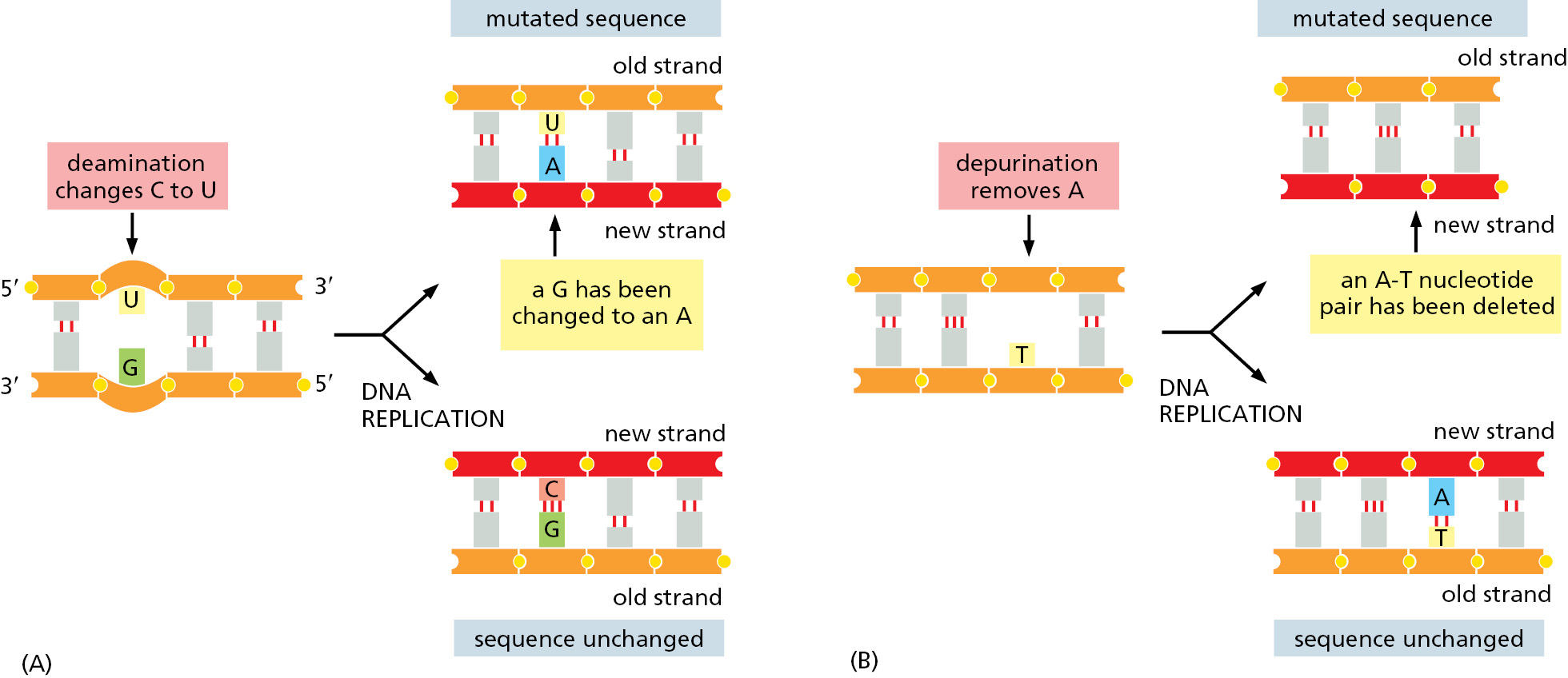
Although DNA is a highly stable material—as required for the storage of genetic information—it is a complex organic molecule that is susceptible, even under normal cell conditions, to spontaneous changes that would lead to mutations if left unrepaired (Figure 5–37 and see Table 5–3). For example, the DNA of each human cell loses about 18,000 purine bases (adenine and guanine) every day because their N-glycosyl linkages to deoxyribose break, a spontaneous hydrolysis reaction called depurination. Similarly, a spontaneous deamination of cytosine to uracil in DNA occurs at a rate of about 100 bases per cell per day (Figure 5–38). DNA bases are also occasionally damaged by encounters with reactive metabolites produced in the cell (for example, the high-energy methyl donor, S-adenosylmethionine) or by exposure to toxic chemicals in the environment. Likewise, ultraviolet radiation from the Sun can produce a covalent linkage between two adjacent pyrimidine bases in DNA to form, for example, thymine dimers (Figure 5–39). If left uncorrected, most of these changes would lead either to the deletion of one or more base pairs or to a base-pair substitution in the daughter DNA chain when the DNA is replicated (Figure 5–40). These mutations would then be propagated throughout all subsequent cell generations. Such a high rate of unrepaired random changes in the DNA sequence would have disastrous consequences, both in the germ line and in somatic tissues.
|
TABLE 5–3 Endogenous DNA Lesions Arising and Repaired in a Diploid Mammalian Cell in 24 Hours |
|
|
DNA lesion |
Number repaired in 24 hr |
|
Hydrolysis |
|
|
Depurination |
18,000 |
|
Depyrimidination |
600 |
|
Cytosine deamination |
100 |
|
5-Methylcytosine deamination |
10 |
|
Oxidation |
|
|
8-oxoguanine |
1500 |
|
Ring-saturated pyrimidines (thymine glycol, cytosine hydrates) |
2000 |
|
Lipid peroxidation products (M1G, etheno-A, etheno-C) |
1000 |
|
Nonenzymatic methylation by S-adenosylmethionine |
|
|
7-Methylguanine |
6000 |
|
3-Methyladenine |
1200 |
|
Nonenzymatic methylation by nitrosated polyamines and peptides |
|
|
C6-Methylguanine |
20–100 |
|
The DNA lesions listed in the table are the result of the normal chemical reactions that take place in cells. Cells that are exposed to external chemicals and radiation suffer greater and more diverse forms of DNA damage. (From T. Lindahl and D.E. Barnes, Cold Spring Harb. Symp. Quant. Biol. 65:127–133, 2000.) |
|
The DNA Double Helix Is Readily Repaired
The double-helical structure of DNA is ideally suited for repair because it carries two separate copies of all the genetic information—one in each of its two strands. Thus, when one strand is damaged, the complementary strand retains an intact copy of the same information, and this copy is generally used as the template to restore the correct nucleotide sequences to the damaged strand.
An indication of the importance of a double-strand helix to the safe storage of genetic information is that all cells use it; only a few small viruses use single-stranded DNA or RNA as their genetic material. The types of repair processes described in this part of the chapter cannot operate on such nucleic acids, and once damaged, the chance of a permanent nucleotide change occurring in these single-strand genomes of viruses is thus very high. It seems that only tiny genomes (and therefore tiny targets for DNA damage) can have their genetic information successfully carried in any molecule other than a DNA double helix.
DNA Damage Can Be Removed by More Than One Pathway
Cells have multiple pathways to repair their DNA using different enzymes that act upon different kinds of lesions. Figure 5–41 shows two of the most common pathways. In both, the damage is excised, the original DNA sequence is restored by a high-fidelity DNA polymerase using the undamaged strand as its template, and the remaining break in the double helix is sealed by DNA ligase (see Figure 5–12).
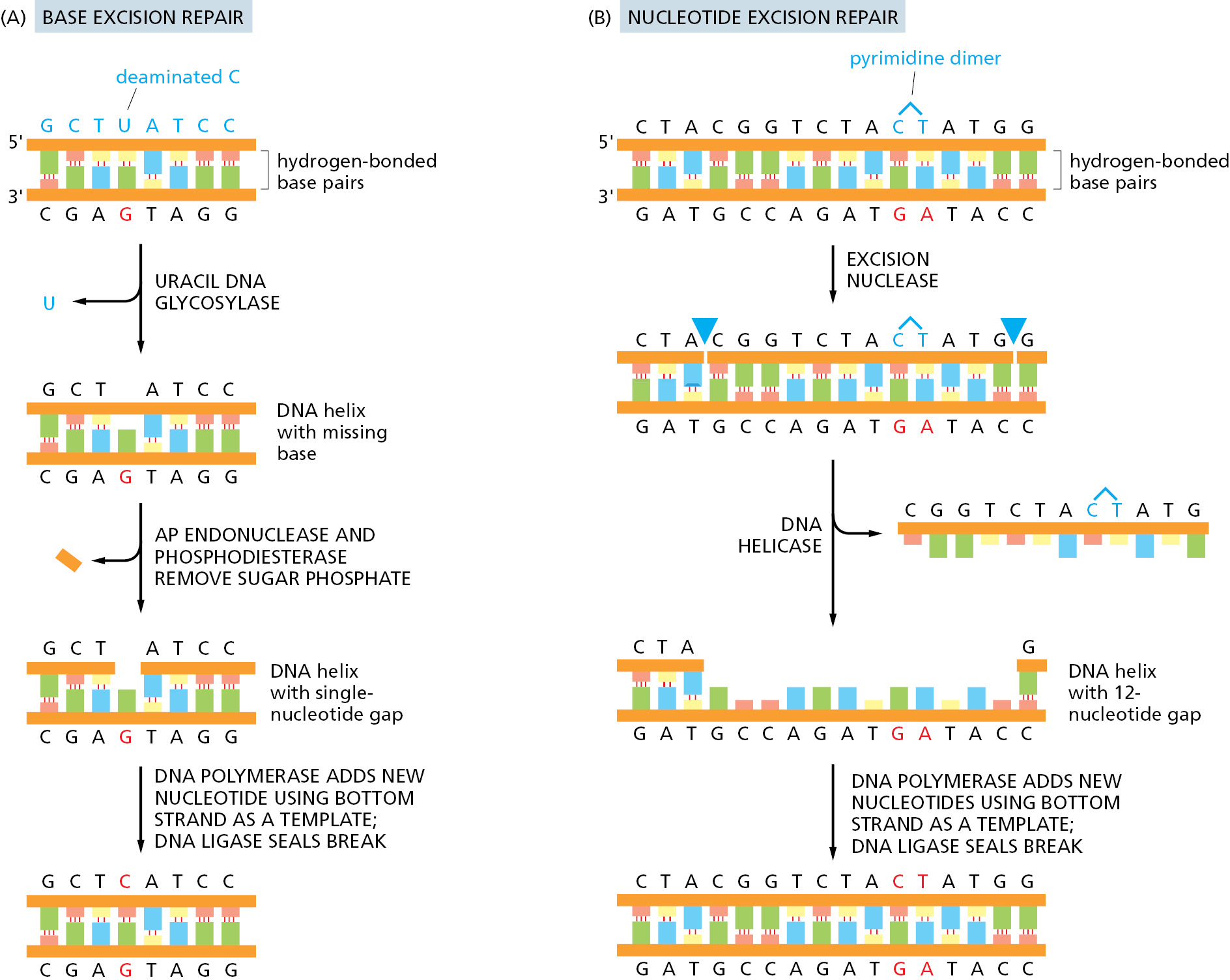
The two pathways differ in the way in which they remove the damage from DNA. The first pathway, called base excision repair, involves a battery of enzymes called DNA glycosylases, each of which can recognize a specific type of altered base in DNA and catalyze its hydrolytic removal from the DNA backbone. There are many types of these enzymes, including those that remove deaminated C’s, deaminated A’s, different types of alkylated or oxidized bases, bases with opened rings, and bases in which a carbon–carbon double bond has been accidentally converted to a carbon–carbon single bond. How are altered bases detected in the double helix? A key step is an enzyme-mediated “flipping-out” of the altered nucleotide from the helix, which allows the DNA glycosylase to probe all faces of the base for damage (Figure 5–42). It is thought that these enzymes travel along DNA using base-flipping to evaluate the status of each base. Once an enzyme finds the damaged base that it recognizes, it removes that base from its sugar.
The “missing tooth” created by DNA glycosylase action is recognized by an enzyme called AP endonuclease (AP for apurinic or apyrimidinic, and endo to signify that the nuclease cleaves within the polynucleotide chain), which cuts the phosphodiester backbone, after which the resulting gap is repaired (see Figure 5–41A). Depurination, which is by far the most frequent type of damage suffered by DNA, also leaves a deoxyribose sugar with a missing base. Depurinations are directly repaired beginning with AP endonuclease, following the bottom half of the pathway in Figure 5–41A.
The second major repair pathway is called nucleotide excision repair. This mechanism can repair the damage caused by almost any large change in the structure of the DNA double helix. Such “bulky lesions” include those created by the covalent reaction of DNA bases with large hydrocarbons (such as the carcinogen benzopyrene, found in tobacco smoke, coal tar, and diesel exhaust), as well as the various pyrimidine dimers (T-T, T-C, and C-C) caused by sunlight. In this pathway, a large multienzyme complex scans the DNA for a distortion in the double helix, rather than for a specific base change. Once it finds a lesion, it cleaves the phosphodiester backbone of the abnormal strand on both sides of the distortion, and a DNA helicase peels away the single-strand oligonucleotide containing the lesion. The large gap produced in the DNA helix is then repaired by DNA polymerase and DNA ligase (see Figure 5–41B).
An alternative to these base and nucleotide excision repair processes is the direct chemical reversal of DNA damage, and this strategy is selectively employed for the rapid removal of certain highly mutagenic or cytotoxic lesions. For example, the lesion O6-methylguanine has its methyl group removed by direct transfer to a cysteine residue in the repair protein itself. Because the repair protein is destroyed in the process, each molecule of it can only be used once. In another example, methyl groups in the lesions 1-methyladenine and 3-methylcytosine are “burned off” by an iron-dependent demethylase, with release of formaldehyde from the methylated DNA and regeneration of the native base.
Coupling Nucleotide Excision Repair to Transcription Ensures That the Cell’s Most Important DNA Is Efficiently Repaired
All of a cell’s DNA is under constant surveillance for damage, and the repair mechanisms we have described act on all parts of the genome. However, cells have a way of directing DNA repair to the DNA sequences that are most needed. They do this by linking RNA polymerase, the enzyme that transcribes DNA into RNA as the first step in gene expression, to the nucleotide excision repair pathway. As discussed above, this repair system can correct many different types of DNA damage. RNA polymerase stalls at DNA lesions and, through the use of coupling proteins, directs the excision repair machinery to those sites, thereby selectively repairing genes that are in current use by the cell. In bacteria, where genes are relatively short, the stalled RNA polymerase can be dissociated from the DNA; the DNA is repaired, and the gene is transcribed again from the beginning. In eukaryotes, where genes can be enormously long, a more complex reaction is used to “back up” the RNA polymerase, repair the damage, and then restart the polymerase.
The importance of transcription-coupled excision repair is seen in people with Cockayne syndrome, which is caused by a defect in this coupling. These individuals suffer from growth retardation, skeletal abnormalities, progressive neural retardation, and severe sensitivity to sunlight. Most of these problems are thought to arise from RNA polymerase molecules that become permanently stalled at sites of DNA damage that lie in important genes.
The Chemistry of the DNA Bases Facilitates Damage Detection
The DNA double helix is well suited for repair. As noted earlier, it contains a backup copy of all genetic information. Equally importantly, the nature of the four bases in DNA makes the distinction between undamaged and damaged bases very clear. For example, every possible deamination event in DNA yields an “unnatural” base, which can be directly recognized and removed by a specific DNA glycosylase. Hypoxanthine, for example, is the simplest purine base capable of pairing specifically with C. But hypoxanthine is not used in DNA, presumably because it is the direct deamination product of A. Instead G, with a second amino group, pairs with C: G cannot form from A by spontaneous deamination, and its own deamination product (xanthine) is likewise unique (Figure 5–43).
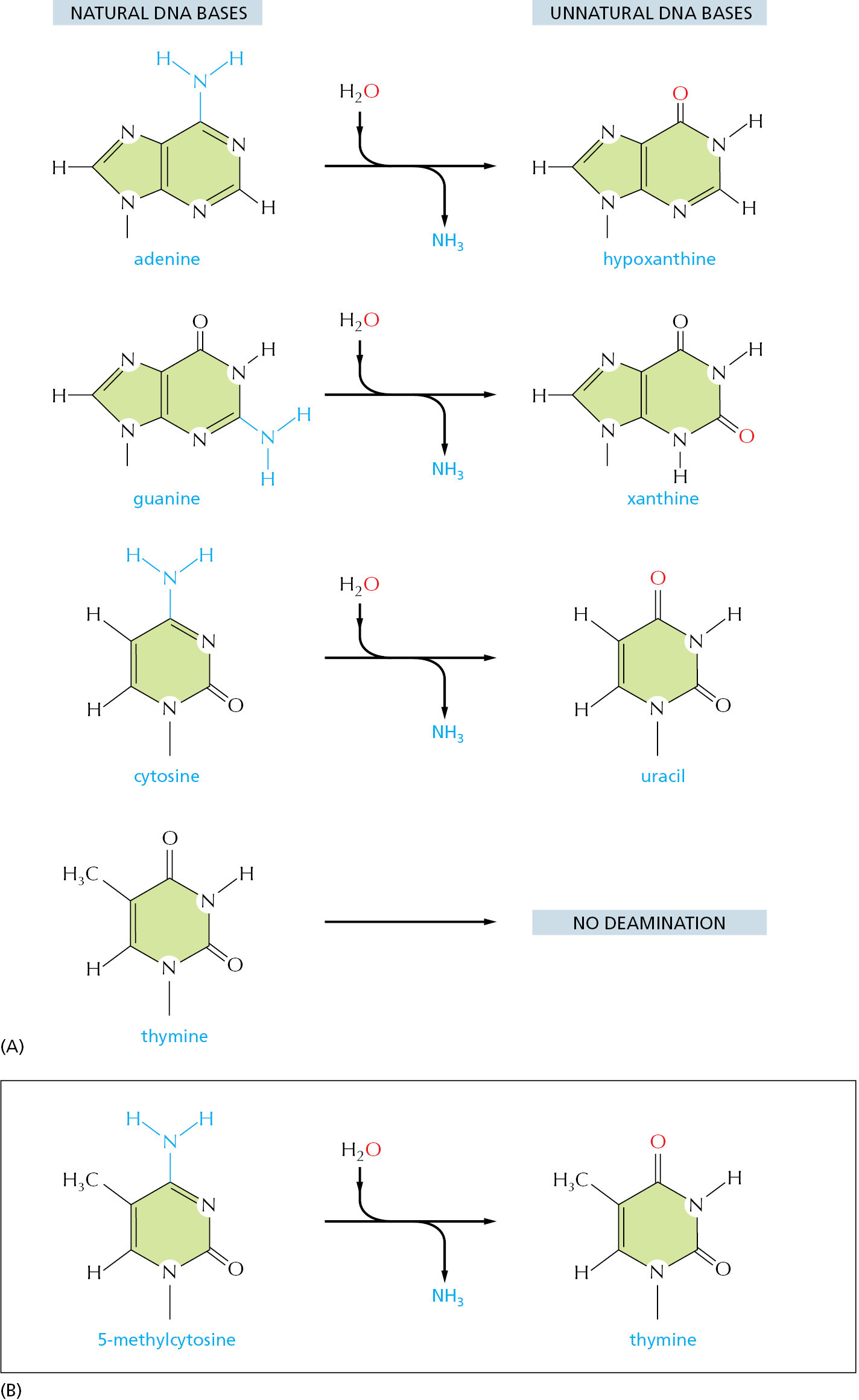
As discussed in Chapter 6, RNA is thought, on an evolutionary time scale, to have served as the genetic material before DNA, and it seems likely that the genetic code was initially carried in the four nucleotides A, C, G, and U. This raises the question of why the U in RNA was replaced in DNA by T (which is 5-methyl U). We have seen that the spontaneous deamination of C converts it to U, but that this event is rendered relatively harmless by uracil DNA glycosylase. However, if DNA contained U as a natural base, the repair system would not be able to distinguish a deaminated C from a naturally occurring U.
A special situation occurs in vertebrate DNA, in which selected C nucleotides are methylated at specific CG sequences that are associated with inactive genes (discussed in Chapter 7). The accidental deamination of these methylated C nucleotides produces the natural nucleotide T (see Figure 5–43B) in a mismatched base pair with a G on the opposite DNA strand. To help in repairing deaminated methylated C nucleotides, a special DNA glycosylase recognizes a mismatched base pair involving T in the sequence T-G and removes the T. This DNA repair mechanism must be relatively ineffective, however, because methylated C nucleotides are exceptionally common sites for mutations in vertebrate DNA. It is striking that, even though only about 3% of the C nucleotides in human DNA are methylated, mutations in these methylated nucleotides account for about one-third of the single-base mutations that have been observed in inherited human diseases.
Special Translesion DNA Polymerases Are Used in Emergencies
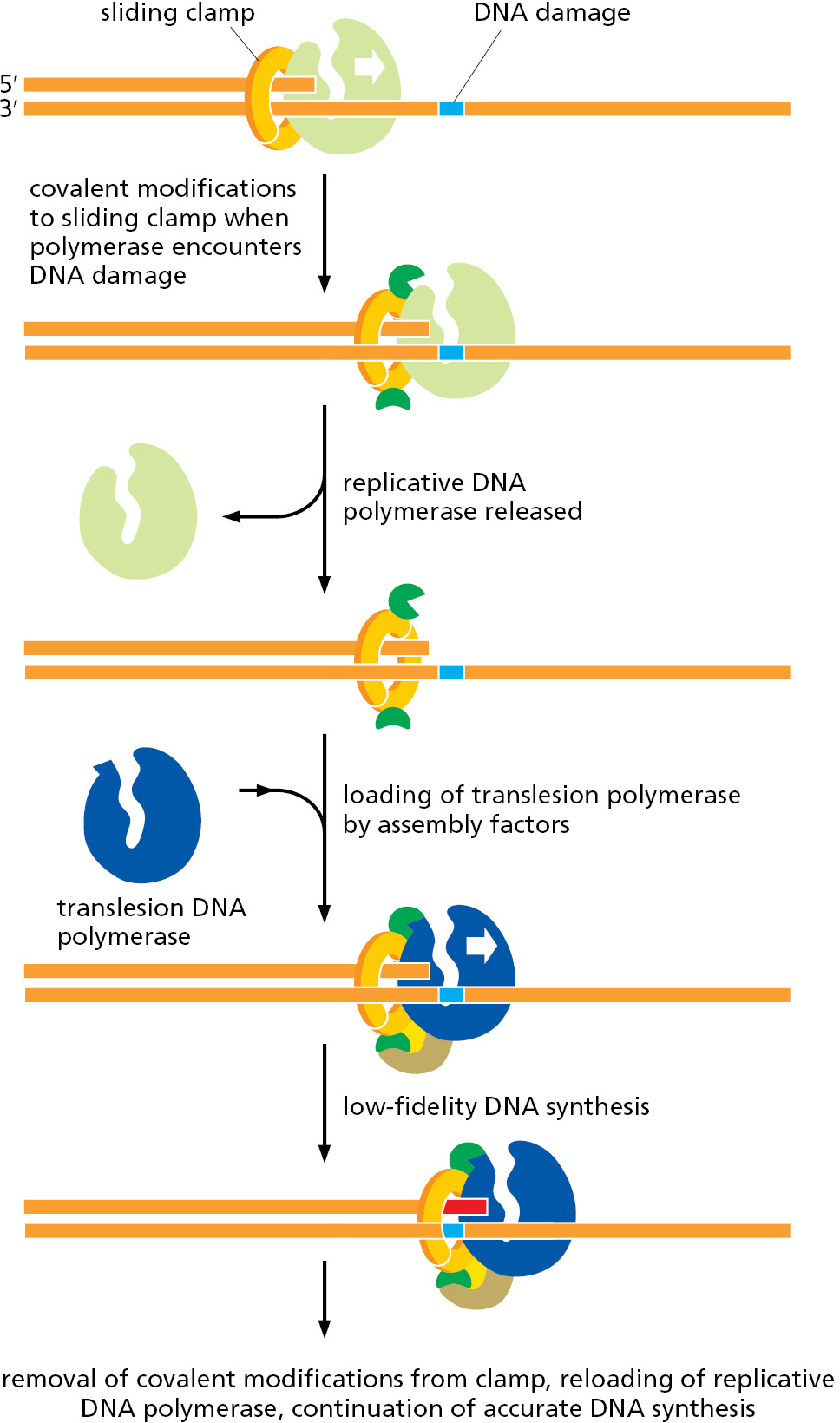
If a cell’s DNA suffers heavy damage, the repair mechanisms that we have discussed are often insufficient to cope with it. In these cases, a different strategy is called into play, one that entails some risk to the cell. The highly accurate replicative DNA polymerases stall when they encounter damaged DNA, and in emergencies cells employ versatile, but less accurate, backup polymerases, known as translesion polymerases, to replicate through the DNA damage.
Human cells contain seven different translesion polymerases, some of which can recognize a specific type of DNA damage and add the nucleotides required to restore the correct sequence. For example, one such polymerase adds two A’s opposite a thymine dimer (see Figure 5–39). Others make only “good guesses,” especially when the template base has been extensively damaged. These enzymes are not as accurate as the normal replicative polymerases even when they copy an undamaged DNA sequence. For one thing, they lack exonucleolytic proofreading activity; in addition, many are much less discriminating than the replicative polymerase in choosing which nucleotide to incorporate initially. Each such translesion polymerase is therefore given a chance to add only one or a few nucleotides before a high-fidelity replicative polymerase resumes DNA synthesis.
Despite their usefulness in allowing heavily damaged DNA to be replicated, these translesion polymerases do, as noted above, pose risks to the cell. They are probably responsible for most of the base-substitution and single-nucleotide deletion mutations that accumulate in genomes. Not only do they frequently produce mutations when copying damaged DNA, they probably also generate mutations—at a low level—on undamaged DNA. Clearly, it is important for the cell to tightly regulate these polymerases, activating them only at sites of DNA damage. Exactly how this happens for each translesion polymerase remains to be discovered, but a conceptual model is presented in Figure 5–44. The same principle applies to many of the DNA repair processes discussed in this chapter: because the enzymes that carry out these reactions are potentially dangerous to the genome, they must be brought into play only at the appropriate damaged sites.
Double-Strand Breaks Are Efficiently Repaired
An especially dangerous type of DNA damage occurs when both strands of the double helix are broken, leaving no intact template strand to enable accurate repair. Ionizing radiation, replication errors, oxidizing agents, and other metabolites produced in the cell cause breaks of this type. If these lesions were left unrepaired, they would quickly lead to the breakdown of chromosomes into smaller fragments and to loss of genes when the cell divides. However, two distinct mechanisms have evolved to deal with this type of damage by restoring an intact double helix: nonhomologous end joining and homologous recombination (Figure 5–45).
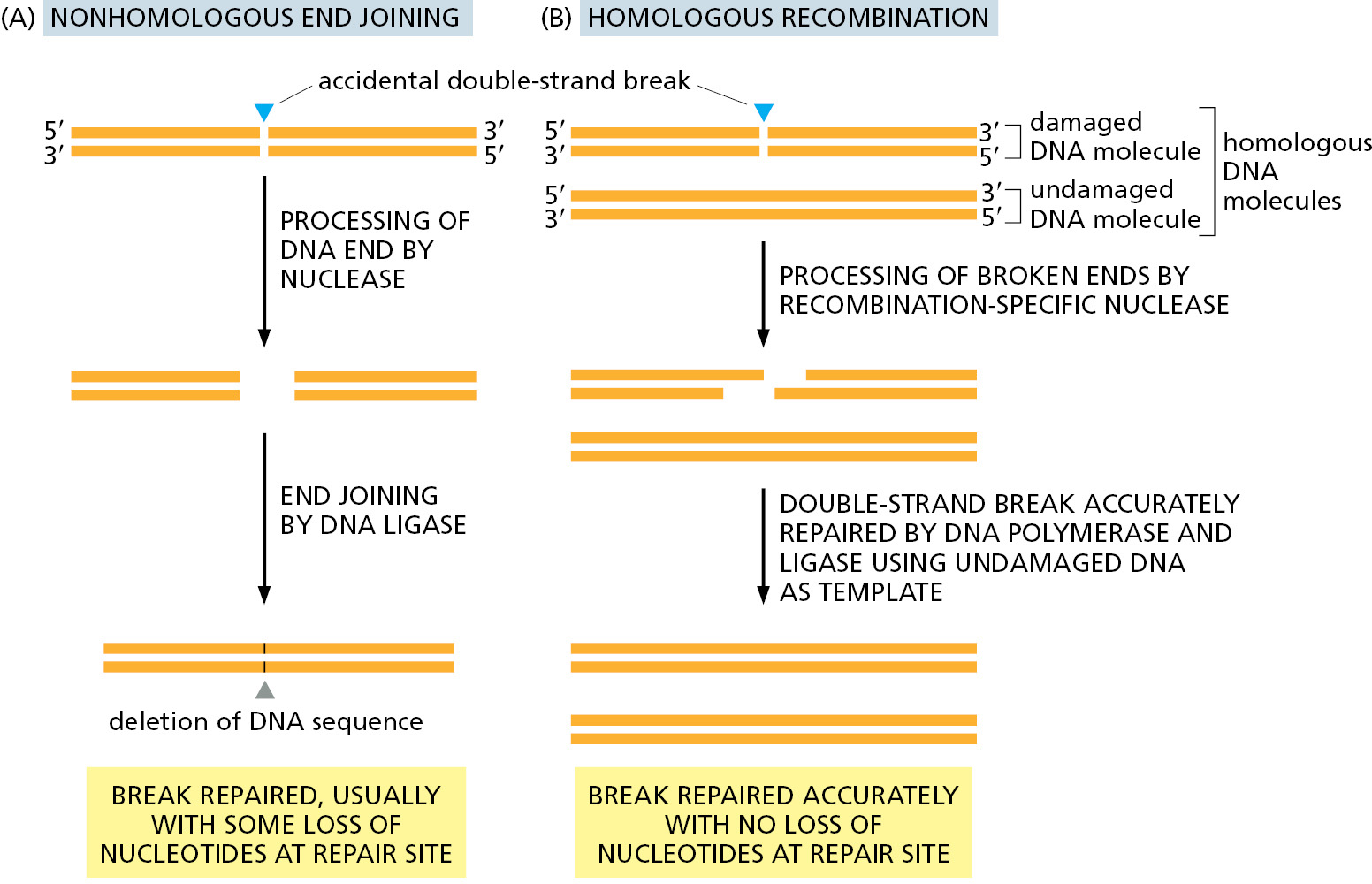
The simplest to understand is nonhomologous end joining, in which the broken ends are processed to remove any damaged nucleotides and simply brought together and rejoined by DNA ligation, generally with the loss of nucleotides at the site of joining (Figure 5–46). This end-joining mechanism, which can be seen as a “quick and dirty” solution to the repair of double-strand breaks, is the predominant way of repairing these lesions in mammalian somatic cells. Although a change in the DNA sequence (a mutation) usually results at the site of breakage, so little of the mammalian genome is essential for life that this mechanism is apparently an acceptable solution to the problem of rejoining broken chromosomes. By the time a human reaches the age of 70, the typical somatic cell contains more than 2000 such “scars,” distributed throughout its genome, representing places where DNA has been inaccurately repaired by nonhomologous end joining.
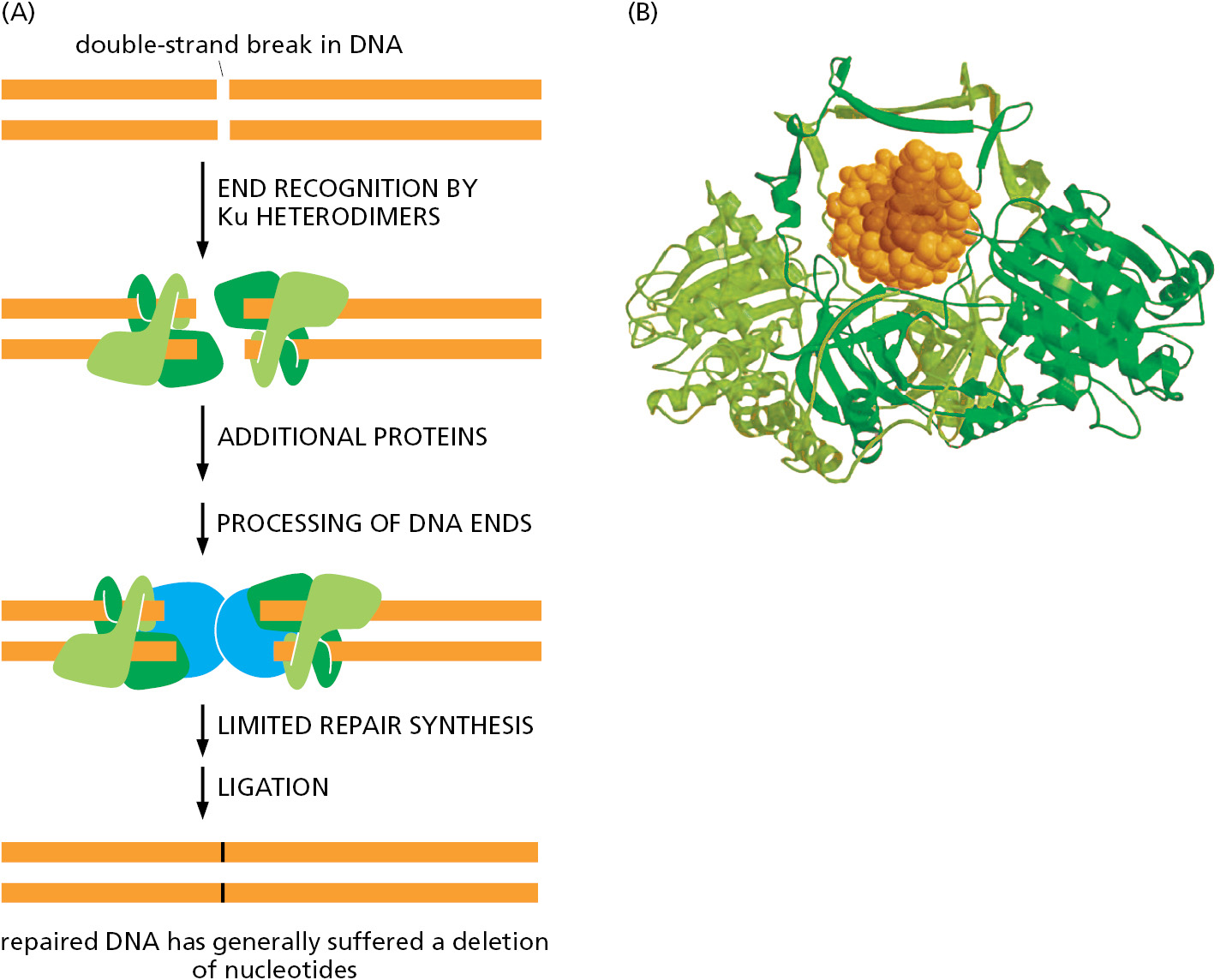
But nonhomologous end joining presents another danger: nonhomologous end joining can occasionally generate rearrangements in which one broken chromosome becomes covalently attached to another. This can result in chromosomes with two centromeres and chromosomes lacking centromeres altogether; both types of aberrant chromosomes are missegregated during cell division. As previously discussed, the specialized structure of telomeres prevents the natural ends of chromosomes from being mistaken for broken DNA and “repaired” in this way.
A much more accurate type of double-strand break repair is also possible (see Figure 5–45B). Here, a damaged DNA molecule is repaired using a second DNA double helix as a template, one with an identical (or nearly identical) DNA sequence. This reaction utilizes homologous recombination, a mechanism to be described later in this chapter. Most organisms employ both nonhomologous end joining and homologous recombination to repair double-strand breaks in DNA. Nonhomologous end joining predominates in humans; homologous recombination is used only in the S and G2 cell-cycle phases, when one newly replicated daughter molecule can act as a template to repair damage to the other daughter that remains nearby.
DNA Damage Delays Progression of the Cell Cycle
We have just seen that cells contain multiple enzyme systems that can recognize and repair many types of DNA damage (Movie 5.7). Because of the importance of maintaining intact, undamaged DNA from generation to generation, eukaryotic cells delay the progression of their cell cycle until DNA repair is complete. As discussed in detail in Chapter 17, the orderly progression of the cell cycle is stopped when damaged DNA is detected, and it restarts only when the damage has been repaired. In mammalian cells, the presence of DNA damage can block entry from G1 phase into S phase, it can slow S phase once it has begun, and it can block the transition from G2 phase to M phase. These delays facilitate DNA repair by providing the time needed for the repair to reach completion.
{kind=link}
DNA damage also results in an increased synthesis of many DNA repair enzymes. This response depends on special signaling proteins that sense DNA damage and synthesize more of the DNA repair enzymes appropriate for the damage. The importance of this mechanism is revealed by the phenotype of humans who are born with defects in the gene that encodes the ATM protein. These individuals have the disease ataxia telangiectasia (AT), the symptoms of which include neurodegeneration, a predisposition to cancer, and genome instability. The ATM protein is a large protein kinase that generates the intracellular signals needed to halt the cell cycle in response to many types of spontaneous DNA damage (see Figure 17–60), and individuals with defects in this protein suffer from the effects of unrepaired DNA lesions.
Summary
Genetic information can be stored stably in DNA sequences only because a large set of DNA repair enzymes continually scans the DNA double helix and replaces any damaged nucleotides. Most types of DNA repair depend on the fact that a DNA molecule carries two copies of its genetic information—one copy on each of its two complementary strands. This allows an accidental lesion on one strand to be removed by a repair enzyme and a corrected strand then resynthesized by reference to the information in the undamaged strand.
Most of the damage to DNA bases is excised by one of two major DNA repair pathways. In base excision repair, the altered base is removed by a DNA glycosylase enzyme, followed by excision of the resulting sugar phosphate. In nucleotide excision repair, a small section of the DNA strand surrounding the damage is removed from the DNA double helix. In both cases, the gap left in the DNA helix is filled in by the sequential action of DNA polymerase and DNA ligase, using the undamaged DNA strand as the template. Some types of DNA damage can be repaired by a different strategy—the direct chemical reversal of the damage—which is carried out by specialized repair proteins. Usually, all such corrections are completed prior to DNA replication. But if not, a special class of inaccurate DNA polymerases, called translesion polymerases, is used to bypass the damage, allowing the cell to survive but sometimes creating permanent mutations at the sites of damage.
Other critical repair systems—based on either nonhomologous end joining or homologous recombination—are needed to reseal the accidental double-strand breaks that occasionally occur in the DNA helix. In most cells, an elevated level of DNA damage causes a delay in the cell cycle, which helps to ensure that the damage is repaired before the cell divides.
Glossary
- DNA repair
- A set of different enzymatic processes for repairing the many accidental lesions that occur continually in DNA.
- base excision repair
- DNA repair pathway in which single faulty bases are removed from the DNA helix and replaced. Compare nucleotide excision repair.
- nonhomologous end joining
- A DNA repair mechanism for rejoining the ends at double-strand breaks in which the two broken ends of DNA are brought together and rejoined by DNA ligation, generally with the loss of one or more nucleotides at the site of joining.
- nucleotide excision repair
- Type of DNA repair that corrects irreversible damage of the DNA double helix, such as that caused by certain chemicals or UV light, by cutting out the damaged region on one strand and resynthesizing it using the undamaged strand as template. Compare base excision repair.