THE SHAPE AND STRUCTURE OF PROTEINS
From a chemical point of view, proteins are by far the most structurally complex and functionally sophisticated molecules known. This is perhaps not surprising, considering that the structure and activity of each protein has developed and been fine-tuned over billions of years of evolution. We start by considering how the position of each amino acid in the long string of amino acids that forms a protein determines its three-dimensional conformation, a shape that is stabilized by noncovalent interactions between different parts of the molecule. Understanding the structure of a protein at the atomic level allows us to see how the precise shape of the protein determines its function.
The Shape of a Protein Is Specified by Its Amino Acid Sequence
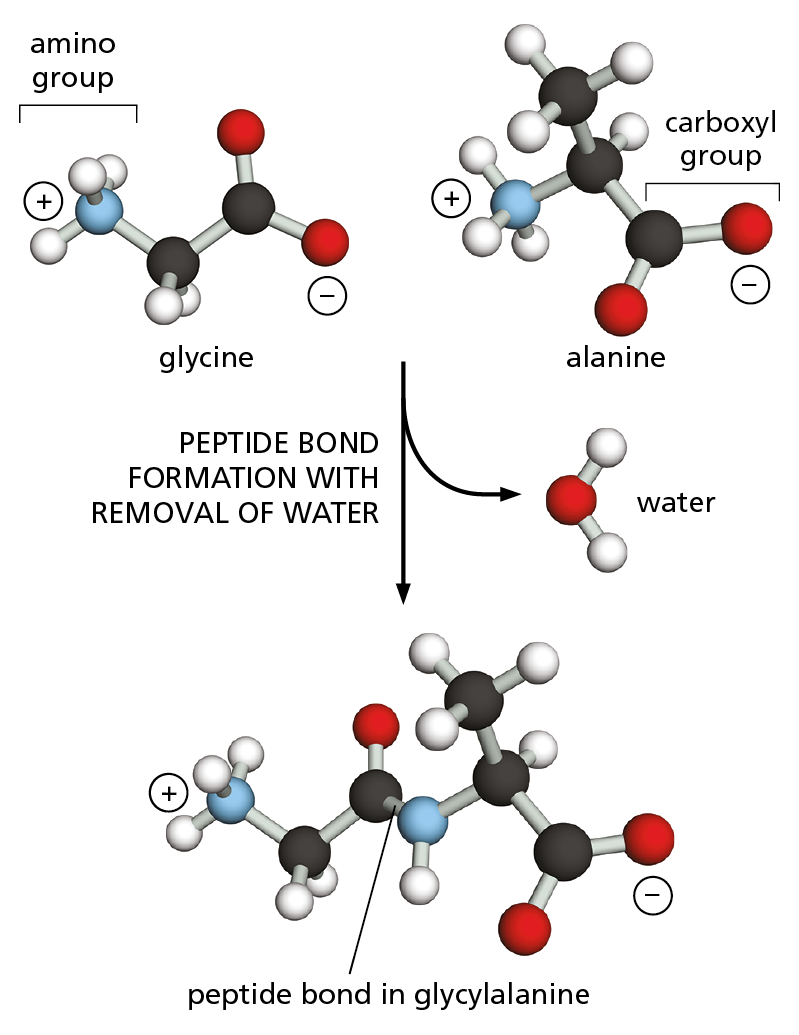
Figure 4–1 Amino acids are linked together by peptide bonds. A covalent peptide bond forms when the carbon atom of the carboxyl group of one amino acid (such as glycine) shares electrons with the nitrogen atom from the amino group of a second amino acid (such as alanine). Because a molecule of water is eliminated, peptide bond formation is classified as a condensation reaction (see Figure 2−31). In this diagram, carbon atoms are black, nitrogen blue, oxygen red, and hydrogen white.
Proteins, as you may recall from Chapter 2, are assembled mainly from a set of 20 different amino acids, each with different chemical properties. A protein molecule is made from a long chain of these amino acids, held together by covalent peptide bonds (Figure 4–1). Proteins are therefore referred to as polypeptides, or polypeptide chains. In each type of protein, the amino acids are present in a unique order, called the amino acid sequence, which is exactly the same from one molecule of that protein to the next. One molecule of human insulin, for example, should have the same amino acid sequence as every other molecule of human insulin. Many thousands of different proteins have been identified, each with its own distinct amino acid sequence.
Each polypeptide chain consists of a backbone that is adorned with a variety of chemical side chains. The polypeptide backbone is formed from a repeating sequence of the core atoms (–N–C–C–) found in every amino acid (Figure 4–2). Because the two ends of each amino acid are chemically different—one sports an amino group (NH3+, also written NH2) and the other a carboxyl group (COO–, also written COOH)—each polypeptide chain has a directionality: the end carrying the amino group is called the amino terminus, or N-terminus, and the end carrying the free carboxyl group is the carboxyl terminus, or C-terminus.
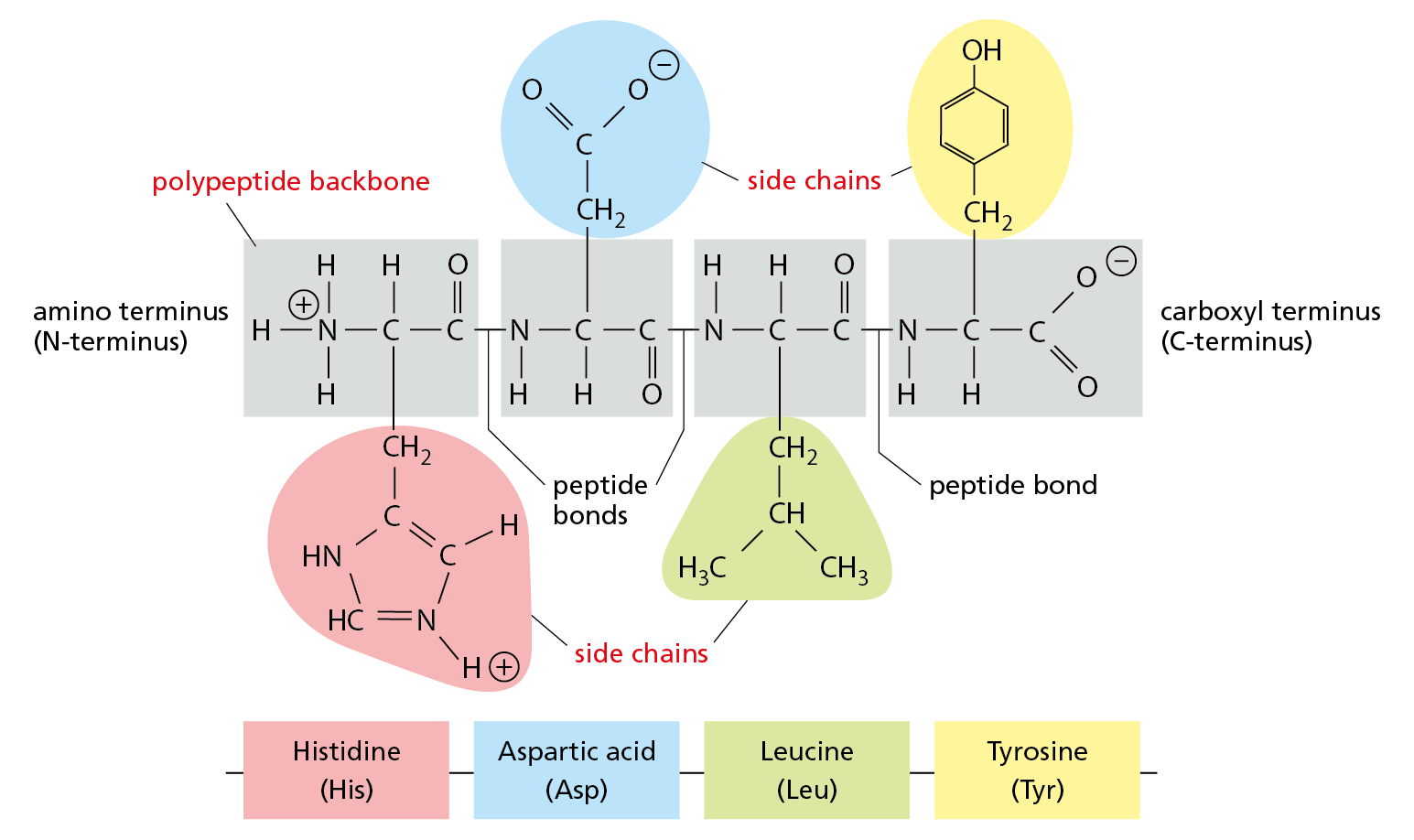
Figure 4–2 A protein is made of amino acids linked together into a polypeptide chain. The amino acids are linked by peptide bonds (see Figure 4–1) to form a polypeptide backbone of repeating structure (gray boxes), from which the side chain of each amino acid projects. The sequence of these chemically distinct side chains—which can be nonpolar (green), polar uncharged (yellow), positively charged (red), or negatively charged (blue)—gives each protein its distinct, individual properties. A small polypeptide of just four amino acids is shown here. Proteins are typically made up of chains of several hundred amino acids, whose sequence is always presented starting with the N-terminus and read from left to right.
Projecting from the polypeptide backbone are the amino acid side chains—the part of the amino acid that is not involved in forming peptide bonds (see Figure 4–2). The side chains give each amino acid its unique properties: some are nonpolar and hydrophobic (“water-fearing”), some are negatively or positively charged, some can be chemically reactive, and so on. The atomic formula for each of the 20 amino acids in proteins is presented in Panel 2–6 (pp. 76–77), and a brief list of the 20 common amino acids, with their abbreviations, is provided in Figure 4–3.
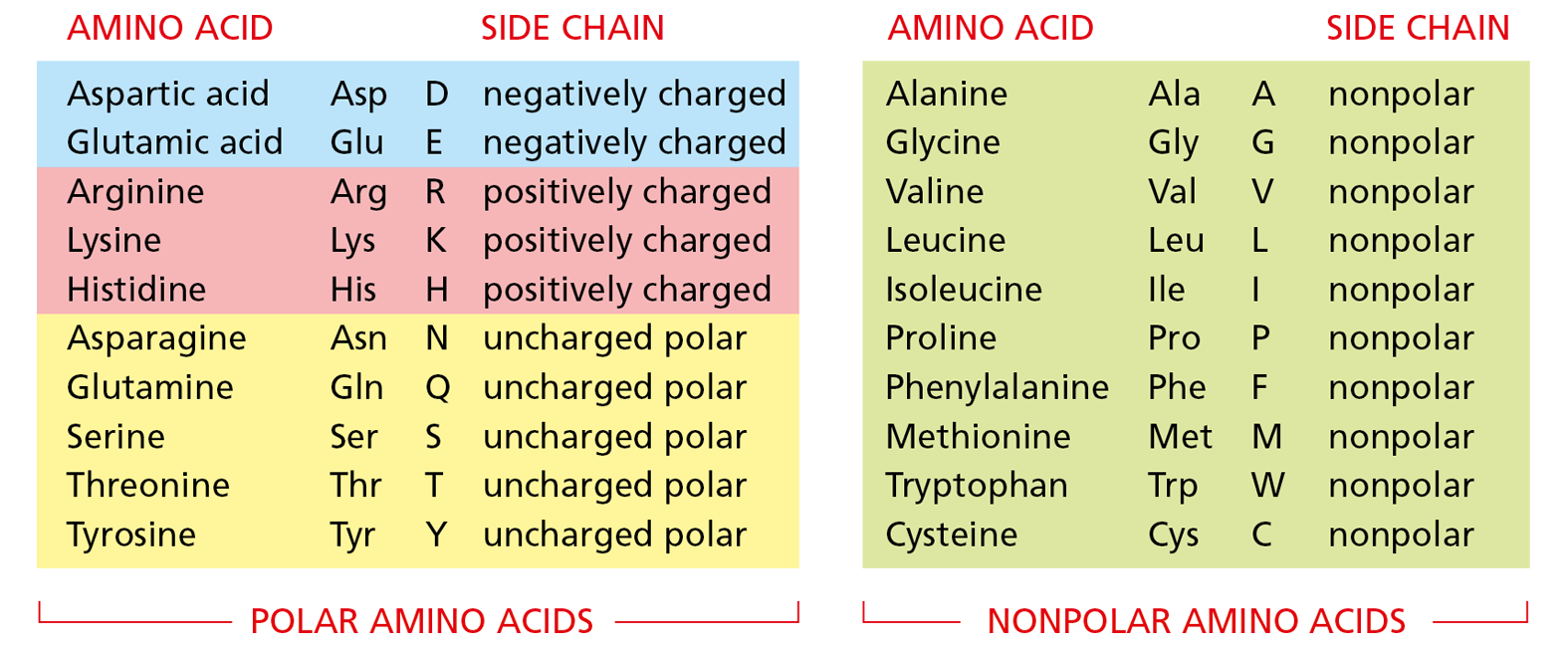
Figure 4–3 Twenty different amino acids are commonly found in proteins. Both three-letter and one-letter abbreviations are given, as well as the character of the side chain. There are equal numbers of polar (hydrophilic) and nonpolar (hydrophobic) side chains, and half of the polar side chains are charged at neutral pH in an aqueous solution. The structures of all of these amino acids are shown in Panel 2−6, pp. 76−77.
Long polypeptide chains are very flexible, as many of the covalent bonds that link the carbon atoms in the polypeptide backbone allow free rotation of the atoms they join. Thus, proteins can in principle fold in an enormous number of ways. The shape of each of these folded chains, however, is constrained by many sets of weak noncovalent bonds that form within proteins. These bonds involve atoms in the polypeptide backbone, as well as atoms within the amino acid side chains. The non-covalent bonds that help proteins fold up and maintain their shape include hydrogen bonds, electrostatic attractions, and van der Waals attractions, which are described in Chapter 2 (see Panel 2–3, pp. 70–71). Because a noncovalent bond is much weaker than a covalent bond, it takes many noncovalent bonds to hold two regions of a polypeptide chain tightly together. The stability of each folded shape is largely determined by the combined strength of large numbers of noncovalent bonds (Figure 4–4).
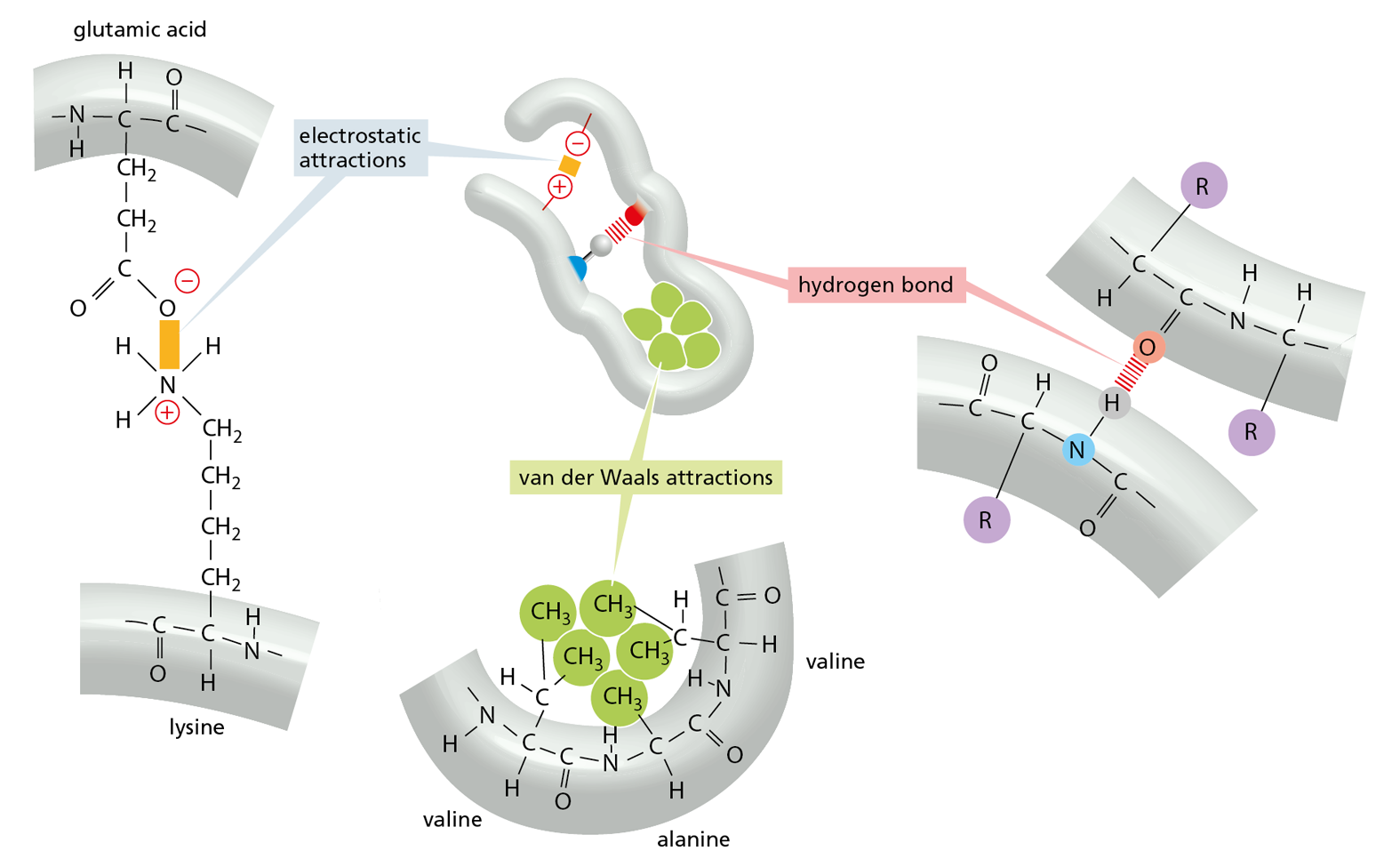
Figure 4–4 Three types of noncovalent bonds help proteins fold. Although a single one of any of these bonds is quite weak, many of them together can create a strong bonding arrangement that stabilizes a particular three-dimensional structure, as in the small polypeptide shown in the center. R is often used as a general designation for an amino acid side chain. Protein folding is also aided by hydrophobic forces, as shown in Figure 4–5.
A fourth weak interaction, the hydrophobic force, also has a central role in determining the shape of a protein. In an aqueous environment, hydrophobic molecules, including the nonpolar side chains of particular amino acids, tend to be forced together to minimize their disruptive effect on the hydrogen-bonded network of the surrounding water molecules (see Panel 2−3, pp. 70–71). Therefore, an important factor governing the folding of any protein is the distribution of its polar and nonpolar amino acids. The nonpolar (hydrophobic) side chains—which belong to amino acids such as phenylalanine, leucine, valine, and tryptophan (see Figure 4–3)—tend to cluster in the interior of the folded protein (just as hydrophobic oil droplets coalesce to form one large drop). Tucked away inside the folded protein, hydrophobic side chains can avoid contact with the aqueous environment that surrounds them inside a cell. In contrast, polar side chains—such as those belonging to arginine, glutamine, and histidine—tend to arrange themselves near the outside of the folded protein, where they can form hydrogen bonds with water and with other polar molecules (Figure 4–5). When polar amino acids are buried within the protein, they are usually hydrogen-bonded to other polar amino acids or to the polypeptide backbone (Figure 4–6).
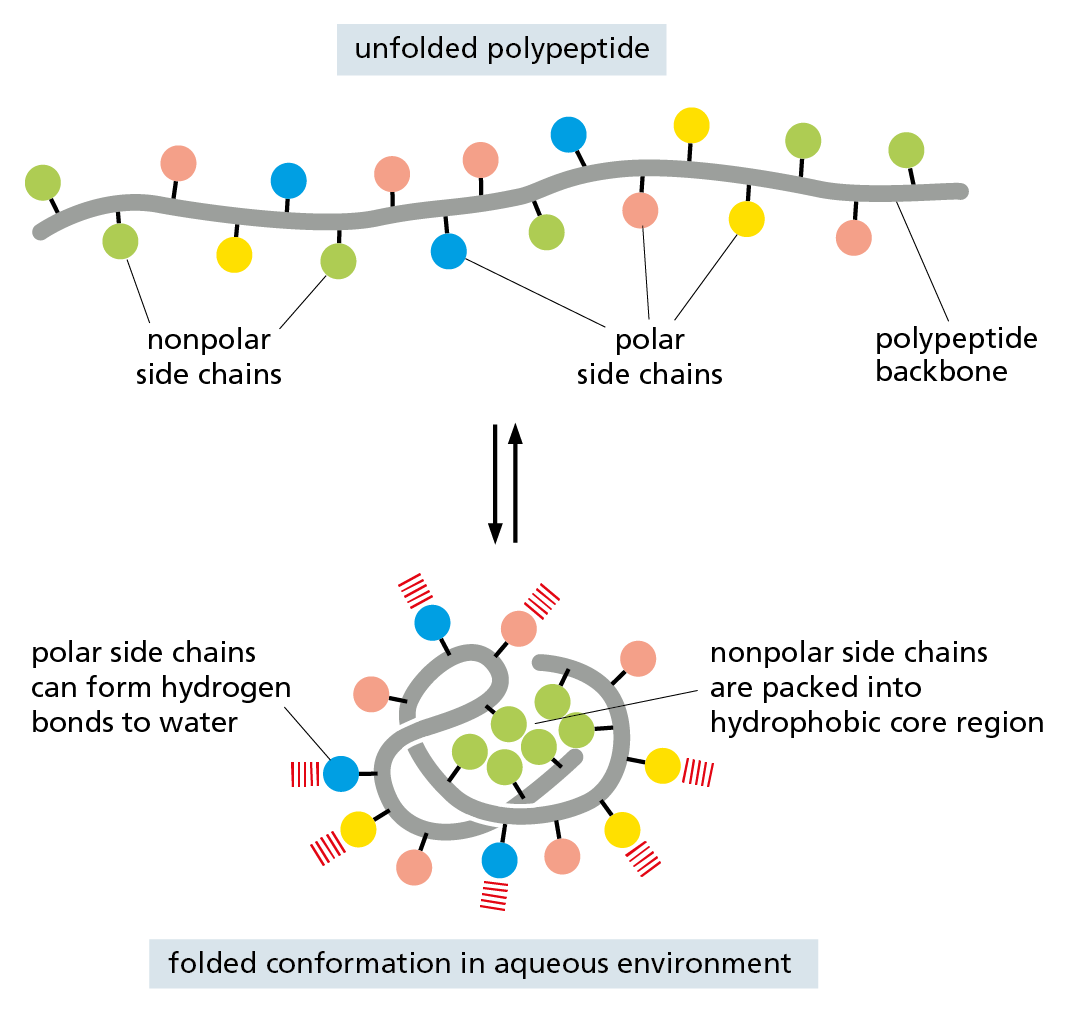
Figure 4–5 Hydrophobic forces help proteins fold into compact conformations. In a folded protein, polar amino acid side chains tend to be displayed on the surface, where they can interact with water; nonpolar amino acid side chains are buried on the inside to form a tightly packed hydrophobic core of atoms that are hidden from water.
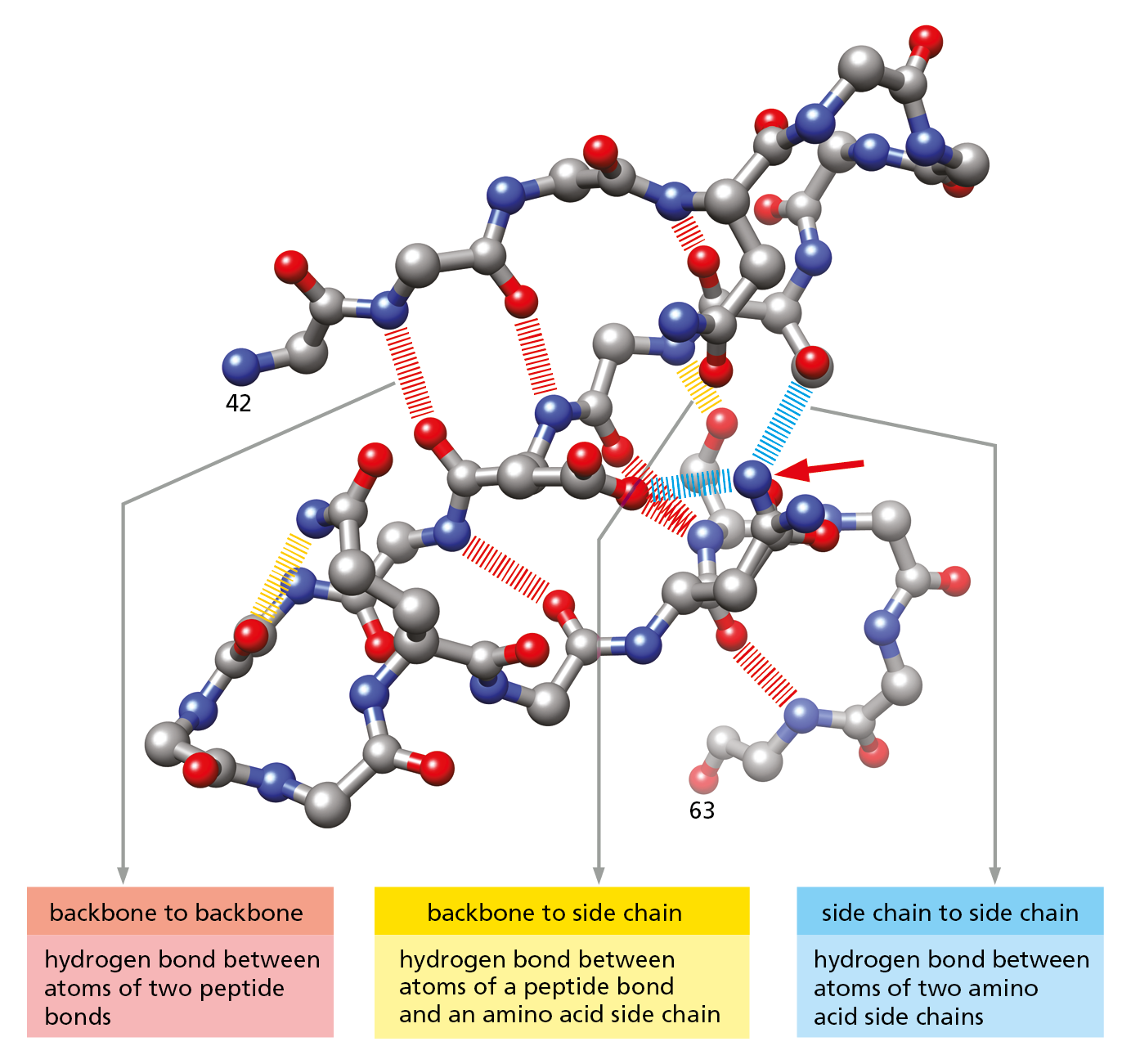
Figure 4–6 Hydrogen bonds within a protein molecule help stabilize its folded shape. Large numbers of hydrogen bonds form between adjacent regions of a folded polypeptide chain. The structure shown is a portion of the enzyme lysozyme, between amino acids 42 and 63. Hydrogen bonds between two atoms in the polypeptide backbone are shown in red ; those between the backbone and a side chain are shown in yellow ; and those between atoms of two side chains are shown in blue. Note that the same amino acid side chain can make multiple hydrogen bonds (red arrow). In this diagram, nitrogen atoms are blue, oxygen atoms are red, and carbon atoms are gray; hydrogen atoms are not shown. (After C.K. Mathews, K.E. van Holde, and K.G. Ahern, Biochemistry, 3rd ed. San Francisco: Benjamin Cummings, 2000.)
Proteins Fold into a Conformation of Lowest Energy
Each type of protein has a particular three-dimensional structure, which is determined by the order of the amino acids in its polypeptide chain. The final folded structure, or conformation, adopted by any polypeptide chain is determined by energetic considerations: a protein generally folds into the shape in which its free energy (G) is minimized. The folding process is thus energetically favorable, as it releases heat and increases the disorder of the universe (see Panel 3−1, pp. 94–95).
Question 4–1
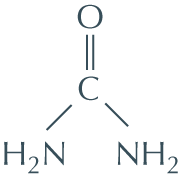
Urea, used in the experiment shown in Figure 4−7, is a molecule that disrupts the hydrogen-bonded network of water molecules. Why might high concentrations of urea unfold proteins? The structure of urea is shown here.
Protein folding has been studied in the laboratory using highly purified proteins. A protein can be unfolded, or denatured, by treatment with solvents that disrupt the noncovalent interactions holding the folded chain together. This treatment converts the protein into a flexible polypeptide chain that has lost its natural shape. Under the right conditions, when the denaturing solvent is removed, the protein often refolds spontaneously into its original conformation—a process called renaturation (Figure 4–7). The fact that a denatured protein can, on its own, refold into the correct conformation indicates that all the information necessary to specify the three-dimensional shape of a protein is contained in its amino acid sequence.
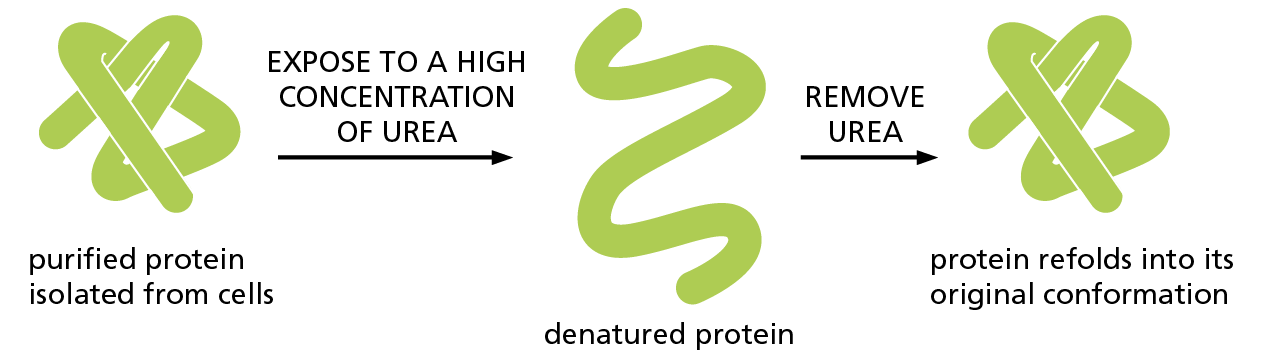
Figure 4–7 Denatured proteins can often recover their natural shapes. This type of experiment demonstrates that the conformation of a protein is determined solely by its amino acid sequence. Renaturation requires the correct conditions and works best for small proteins.
Although a protein chain can fold into its correct conformation without outside help, protein folding in a living cell is generally assisted by a large set of special proteins called chaperone proteins. Some of these chaperones bind to partly folded chains and help them to fold along the most energetically favorable pathway (Figure 4–8). Others form “isolation chambers” in which single polypeptide chains can fold without the risk of forming aggregates in the crowded conditions of the cytoplasm (Figure 4–9). In either case, the final three-dimensional shape of the protein is still specified by its amino acid sequence; chaperones merely make the folding process more efficient and reliable.
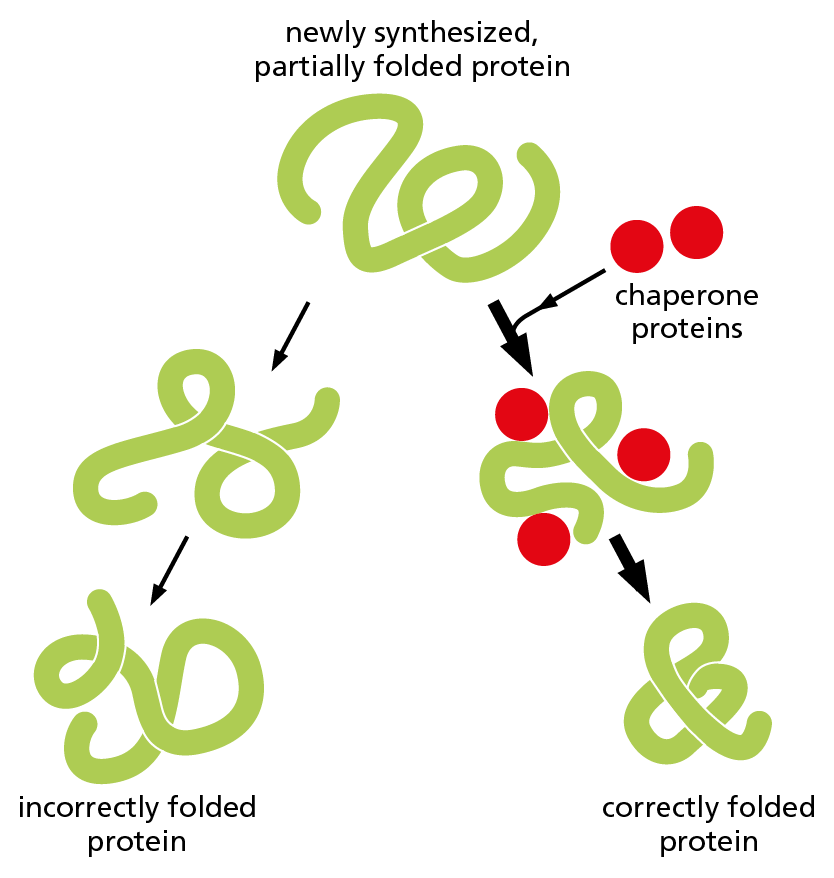
Figure 4–8 Chaperone proteins can guide the folding of a newly synthesized polypeptide chain. The chaperones bind to newly synthesized or partially folded chains and help them to fold along the most energetically favorable pathway. The function of these chaperones requires ATP binding and hydrolysis.
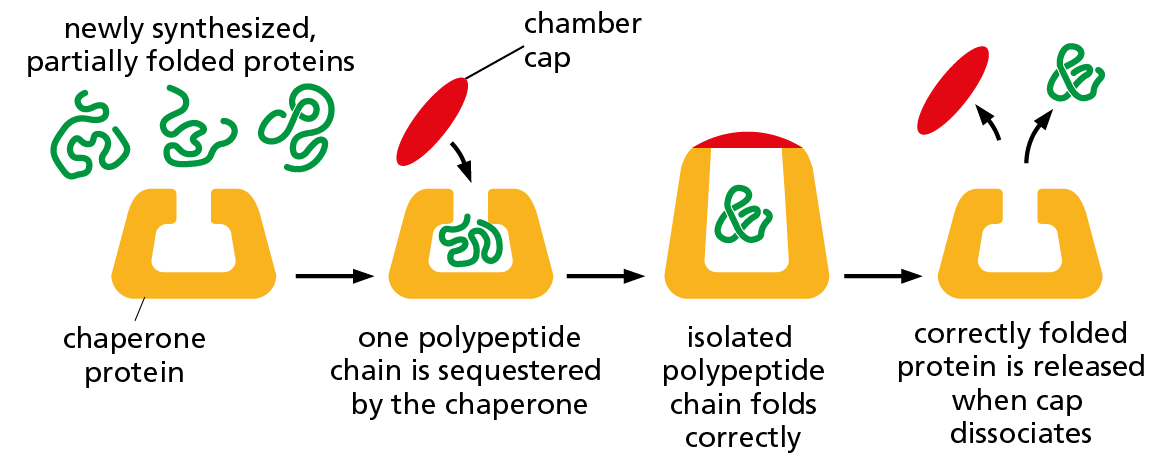
Figure 4–9 Some chaperone proteins act as isolation chambers that help a polypeptide fold. In this case, the barrel of the chaperone provides an enclosed chamber in which a newly synthesized polypeptide chain can fold without the risk of aggregating with other polypeptides in the crowded conditions of the cytoplasm. This system also requires an input of energy from ATP hydrolysis, mainly for the association and subsequent dissociation of the cap that closes off the chamber.
Each protein normally folds into a single, stable conformation. This conformation, however, often changes slightly when the protein interacts with other molecules in the cell. Such changes in shape are crucial to the function of the protein, as we discuss later.
Proteins Come in a Wide Variety of Complicated Shapes
Proteins are the most structurally diverse macromolecules in the cell. Although they range in size from about 30 amino acids to more than 10,000, the vast majority are between 50 and 2000 amino acids long. Proteins can be globular or fibrous, and they can form filaments, sheets, rings, or spheres (Figure 4−10). We will encounter many of these structures throughout the book.
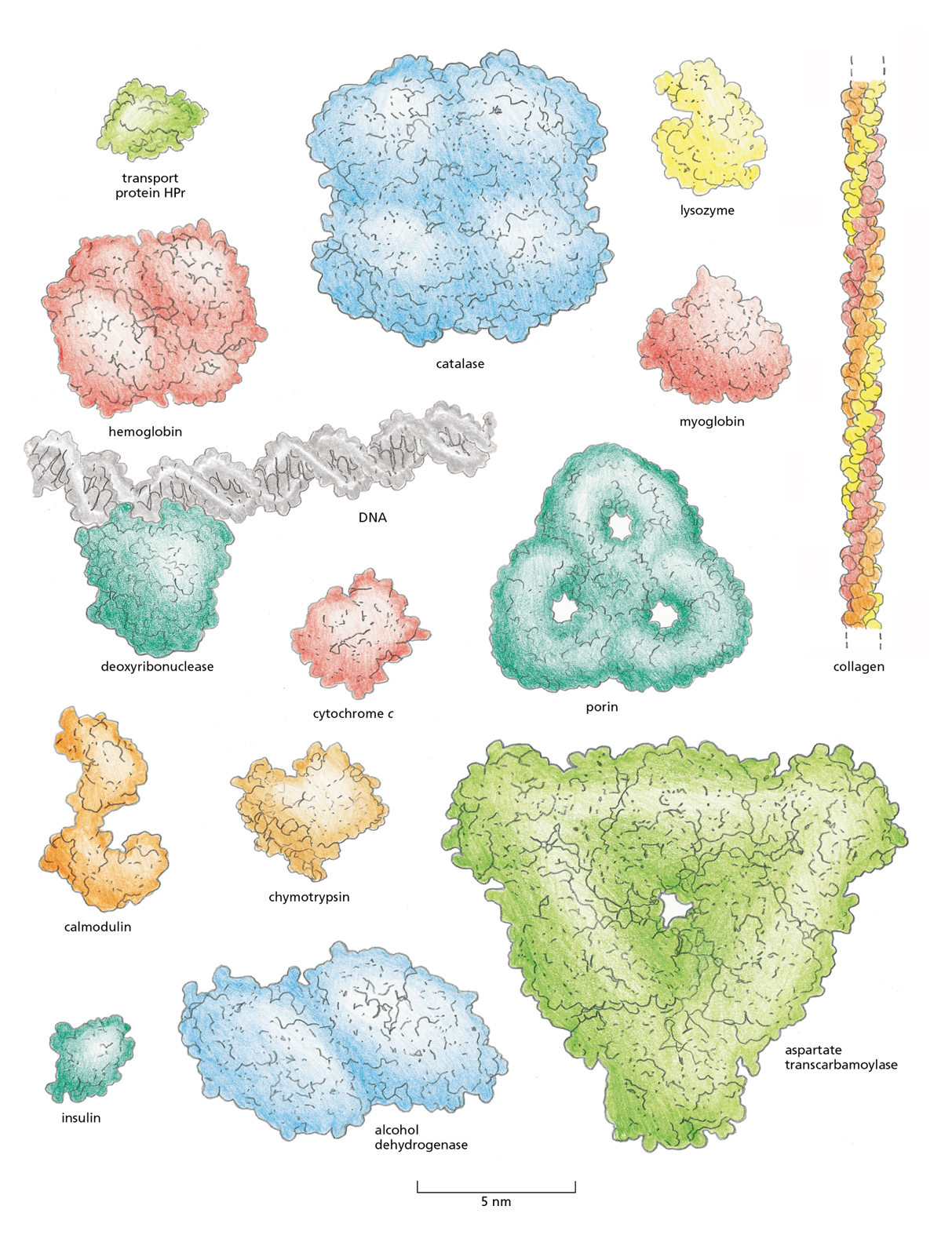
Figure 4−10 Proteins come in a wide variety of shapes and sizes. Each folded polypeptide is shown as a space-filling model, represented at the same scale. In the top-left corner is HPr, the small transport protein featured in detail in Figure 4−11. The protein deoxyribonuclease is shown bound to a portion of a DNA molecule (gray) for comparison.
To date, the structures of about 100,000 different proteins have been determined (using techniques we discuss later in the chapter). Most proteins have a three-dimensional conformation so intricate and irregular that their structure would require the rest of the chapter to describe in detail. But we can get some sense of the intricacies of polypeptide structure by looking at the conformation of a relatively small protein, such as the bacterial transport protein HPr.
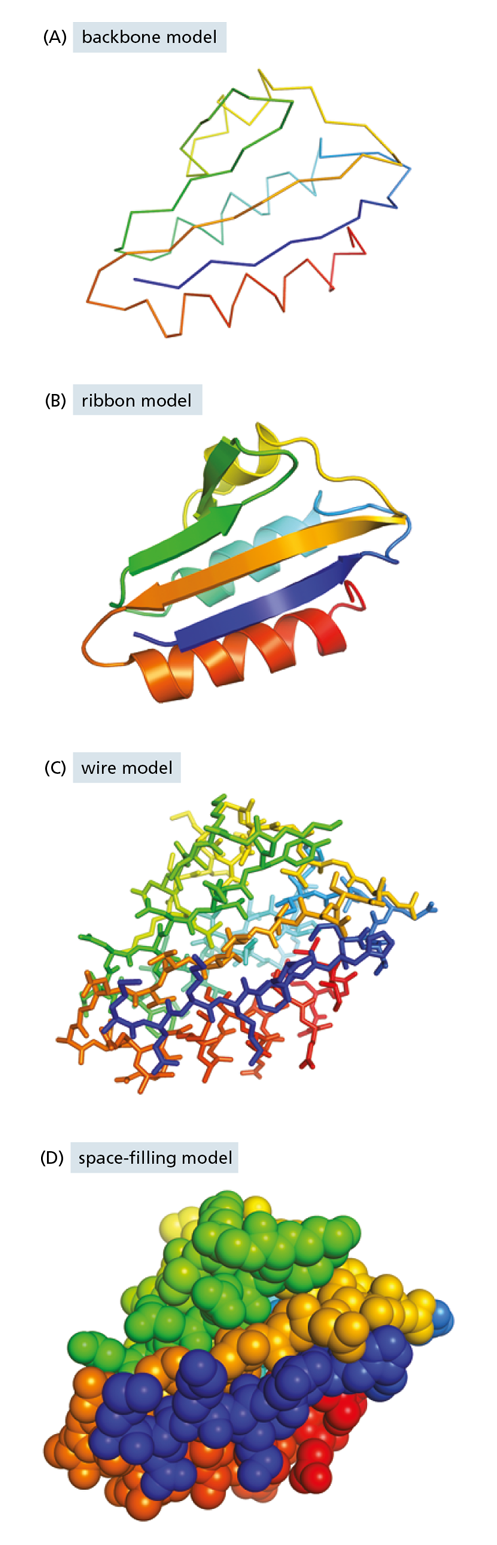
Figure 4−11 Protein conformation can be represented in a variety of ways. Shown here is the structure of the small bacterial transport protein HPr. The images are colored to make it easier to trace the path of the polypeptide chain. In these models, the region of polypeptide chain carrying the protein’s N-terminus is purple and that near its C-terminus is red.
This small protein, only 88 amino acids long, facilitates the transport of sugar into bacterial cells. In Figure 4−11, we present HPr’s three-dimensional structure in four different ways, each of which emphasizes different features of the protein. The backbone model (see Figure 4−11A) shows the overall organization of the polypeptide chain and provides a straightforward way to compare the structures of related proteins. The ribbon model (see Figure 4−11B) shows the polypeptide backbone in a way that emphasizes its most conspicuous folding patterns, which we describe in detail shortly. The wire model (see Figure 4−11C) includes the positions of all the amino acid side chains; this view is especially useful for predicting which amino acids might be involved in the protein’s activity. Finally, the space-filling model (see Figure 4−11D) provides a contour map of the protein surface, which reveals which amino acids are exposed on the surface and shows how the protein might look to a small molecule such as water or to another macromolecule in the cell.
The structures of larger proteins—or of multiprotein complexes—are even more complicated. To visualize such detailed and intricate structures, scientists have developed various computer-based tools to emphasize different features of a protein, only some of which are depicted in Figure 4–11. All of these images can be displayed on a computer screen and readily rotated and magnified to view all aspects of the structure (Movie 4.1).
When the three-dimensional structures of many different protein molecules are compared, it becomes clear that, although the overall conformation of each protein is unique, some regular folding patterns can be detected, as we discuss next.
The α Helix and the β Sheet Are Common Folding Patterns
More than 60 years ago, scientists studying hair and silk discovered two regular folding patterns that are present in many different proteins. The first to be discovered, called the α helix, was found in the protein α-keratin, which is abundant in skin and its derivatives—such as hair, nails, and horns. Within a year of that discovery, a second folded structure, called a β sheet, was found in the protein fibroin, the major constituent of silk. (Biologists often use Greek letters to name their discoveries, with the first example receiving the designation α, the second β, and so on.)
These two folding patterns are particularly common because they result from hydrogen bonds that form between the N–H and C=O groups in the polypeptide backbone (see Figure 4−6). Because the amino acid side chains are not involved in forming these hydrogen bonds, α helices and β sheets can be generated by many different amino acid sequences. In each case, the protein chain adopts a regular, repeating form. These structural features, and the shorthand cartoon symbols that are often used to represent them in models of protein structures, are presented in Figures 4−12 and 4−13.
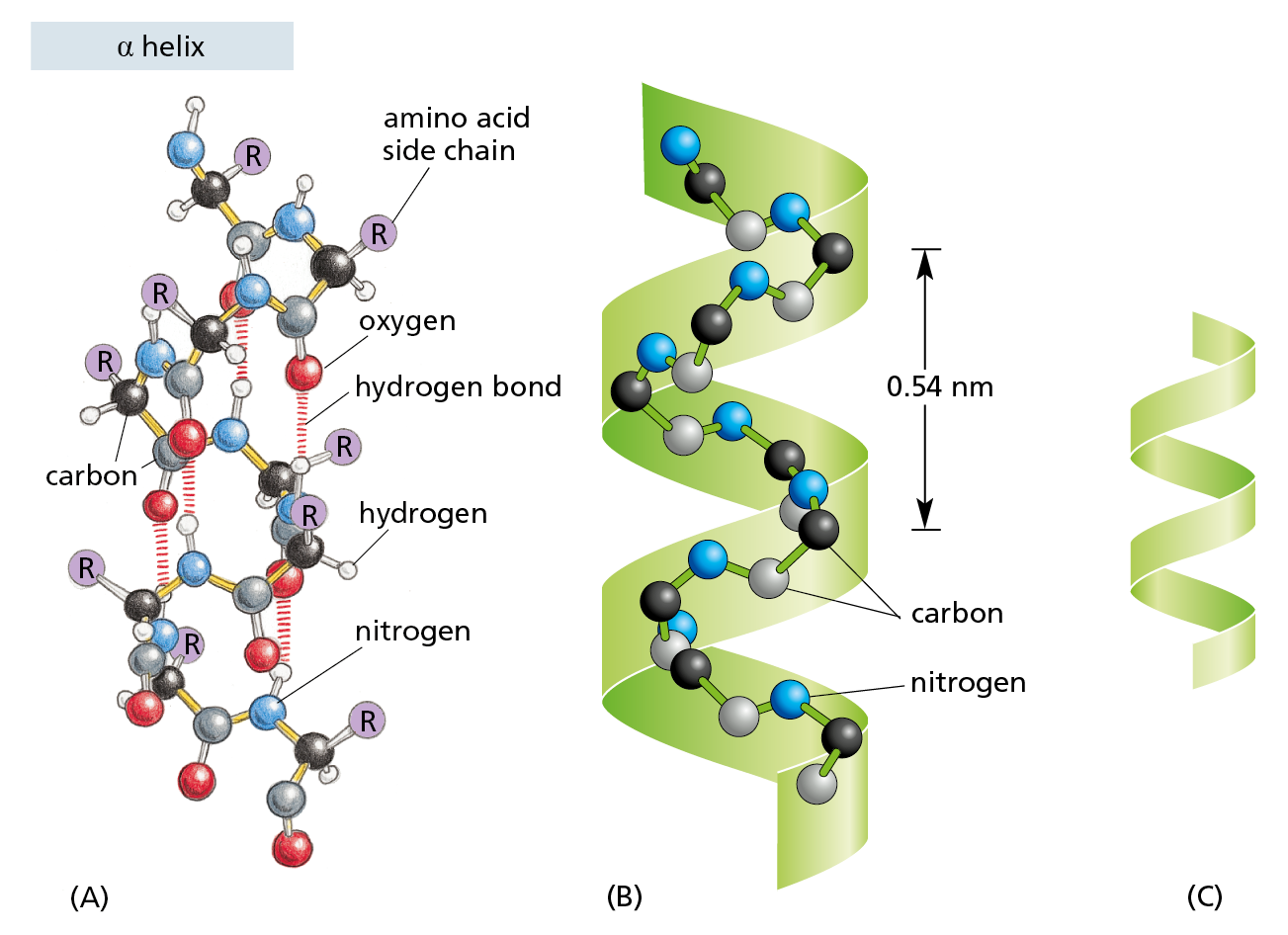
Figure 4−12 Some polypeptide chains fold into an orderly repeating form known as an α helix. (A) In an α helix, the N–H of every peptide bond is hydrogen-bonded to the C=O of a neighboring peptide bond located four amino acids away in the same chain. All of the atoms in the polypeptide backbone are shown; the amino acid side chains are denoted by R. (B) The same polypeptide, showing only the carbon (black and gray) and nitrogen (blue) atoms. (C) Cartoon symbol used to represent an α helix in ribbon models of proteins (see Figure 4−11B).
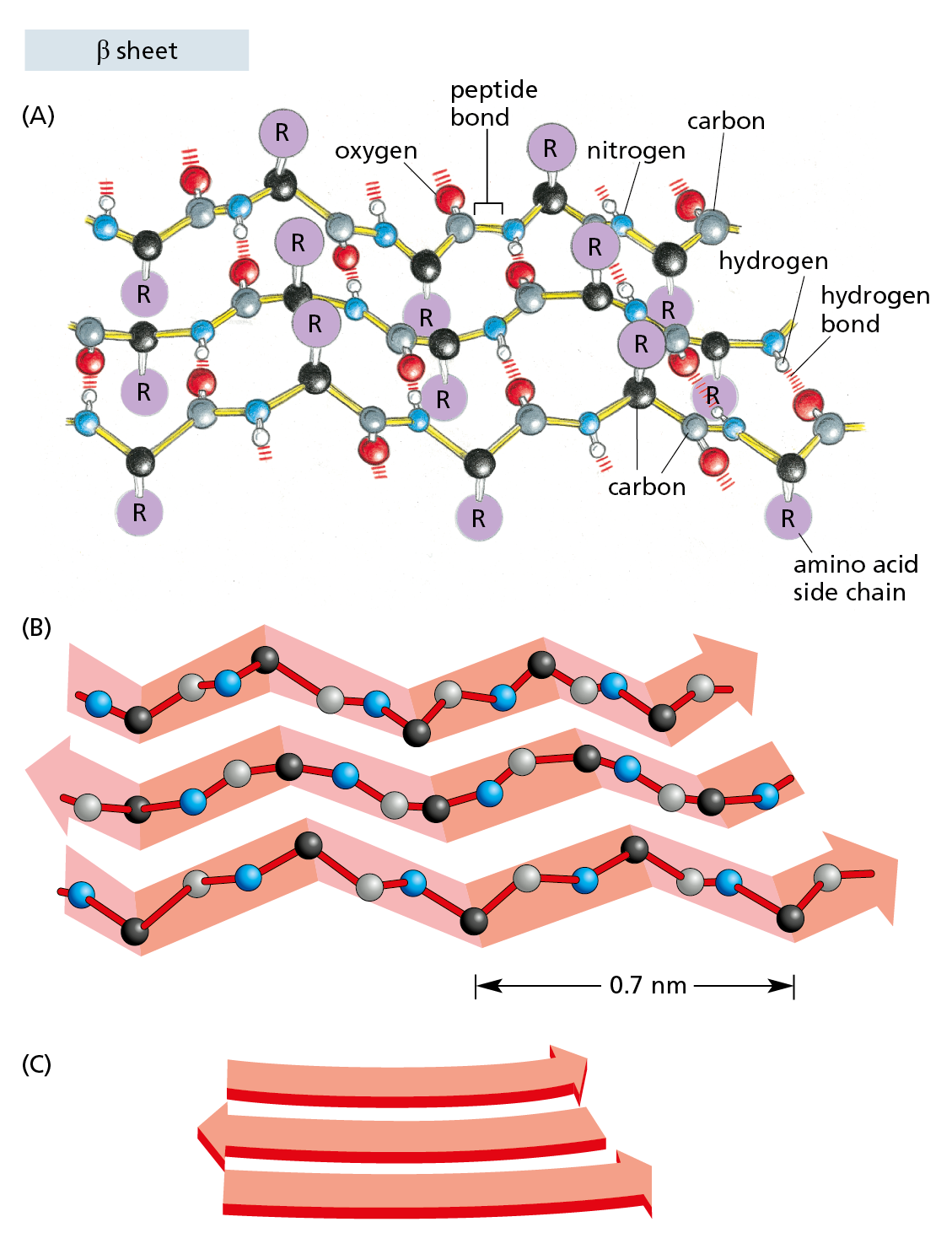
Figure 4−13 Some polypeptide chains fold into an orderly pattern called a β sheet. (A) In a β sheet, several segments (strands) of an individual polypeptide chain are held together by hydrogen-bonding between peptide bonds in adjacent strands. The amino acid side chains in each strand project alternately above and below the plane of the sheet. In the example shown, the adjacent chains run in opposite directions, forming an antiparallel β sheet. All of the atoms in the polypeptide backbone are shown; the amino acid side chains are denoted by R. (B) The same polypeptide, showing only the carbon (black and gray) and nitrogen (blue) atoms. (C) Cartoon symbol used to represent β sheets in ribbon models of proteins (see Figure 4−11B).
Helices Form Readily in Biological Structures
The abundance of helices in proteins is, in a way, not surprising. A helix is generated simply by placing many similar subunits next to one another, each in the same strictly repeated relationship to the one before. Because it is very rare for subunits to join up in a straight line, this arrangement will generally result in a structure that resembles a spiral staircase (Figure 4−14). Depending on the way it twists, a helix is said to be either right-handed or left-handed (see Figure 4−14E). Handedness is not affected by turning the helix upside down, but it is reversed if the helix is reflected in a mirror.
Question 4–2
Remembering that the amino acid side chains projecting from each polypeptide backbone in a β sheet point alternately above and below the plane of the sheet (see Figure 4−13A), consider the following protein sequence: Leu-Lys-Val-Asp-Ile-Ser-Leu-Arg-Leu-Lys-Ile-Arg-Phe-Glu. Do you find anything remarkable about the arrangement of the amino acids in this sequence when incorporated into a β sheet? Can you make any predictions as to how the β sheet might be arranged in a protein? (Hint: consult the properties of the amino acids listed in Figure 4−3.)
An α helix is generated when a single polypeptide chain turns around itself to form a structurally rigid cylinder. A hydrogen bond is made between every fourth amino acid, linking the C=O of one peptide bond to the N–H of another (see Figure 4−12A). This pattern gives rise to a regular right-handed helix with a complete turn every 3.6 amino acids (Movie 4.2).
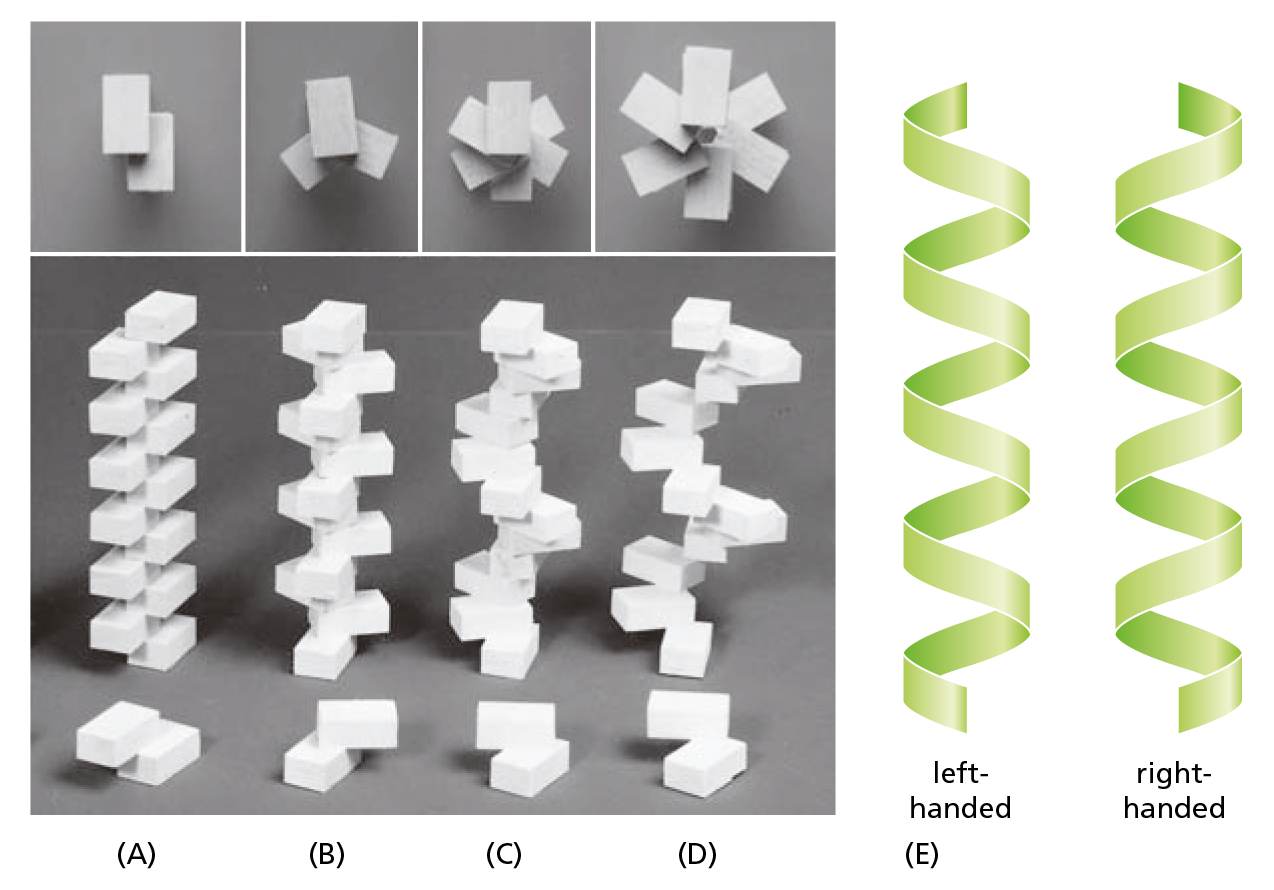
Figure 4−14 A helix is a common, regular, biological structure. A helix will form when a series of similar subunits bind to each other in a regular way. At the bottom, the interaction between two subunits is shown; behind them are the helices that result. These helices have (A) two, (B) three, or (C and D) six subunits per helical turn. At the top, the arrangement of subunits has been photographed from directly above the helix. Note that the helix in (D) has a wider path than that in (C), but the same number of subunits per turn. (E) A helix can be either right-handed or left-handed. As a reference, it is useful to remember that standard metal screws, which advance when turned clockwise, are right-handed. So to judge the handedness of a helix, imagine screwing it into a wall. Note that a helix preserves the same handedness when it is turned upside down. In proteins, α helices are almost always right-handed.
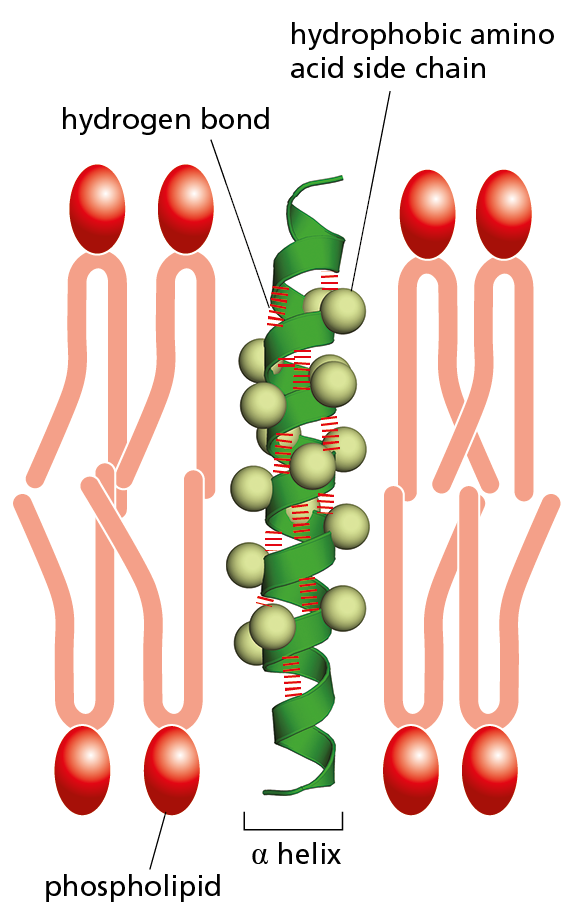
Figure 4–15 Many membrane-bound proteins cross the lipid bilayer as an α helix. The hydrophobic side chains of the amino acids that form the α helix make contact with the hydrophobic hydrocarbon tails of the phospholipid molecules, while the hydrophilic parts of the polypeptide backbone form hydrogen bonds with one another along the interior of the helix. About 20 amino acids are required to span a membrane in this way. Note that, despite the appearance of a space along the interior of the helix in this schematic diagram, the helix is not a channel: no ions or small molecules can pass through it.
Short regions of α helix are especially abundant in proteins that are embedded in cell membranes, such as transport proteins and receptors. We see in Chapter 11 that the portions of a transmembrane protein that cross the lipid bilayer usually form an α helix, composed largely of amino acids with nonpolar side chains. The polypeptide backbone, which is hydrophilic, is hydrogen-bonded to itself inside the α helix, where it is shielded from the hydrophobic lipid environment of the membrane by the protruding nonpolar side chains (Figure 4−15).
Sometimes two (or three) α helices will wrap around one another to form a particularly stable structure called a coiled-coil. This structure forms when the α helices have most of their nonpolar (hydrophobic) side chains along one side, so they can twist around each other with their hydrophobic side chains facing inward—minimizing contact with the aqueous cytosol (Figure 4−16). Long, rodlike coiled-coils form the structural framework for many elongated proteins, including the α-keratin found in hair and the outer layer of the skin, as well as myosin, the motor protein responsible for muscle contraction (discussed in Chapter 17).
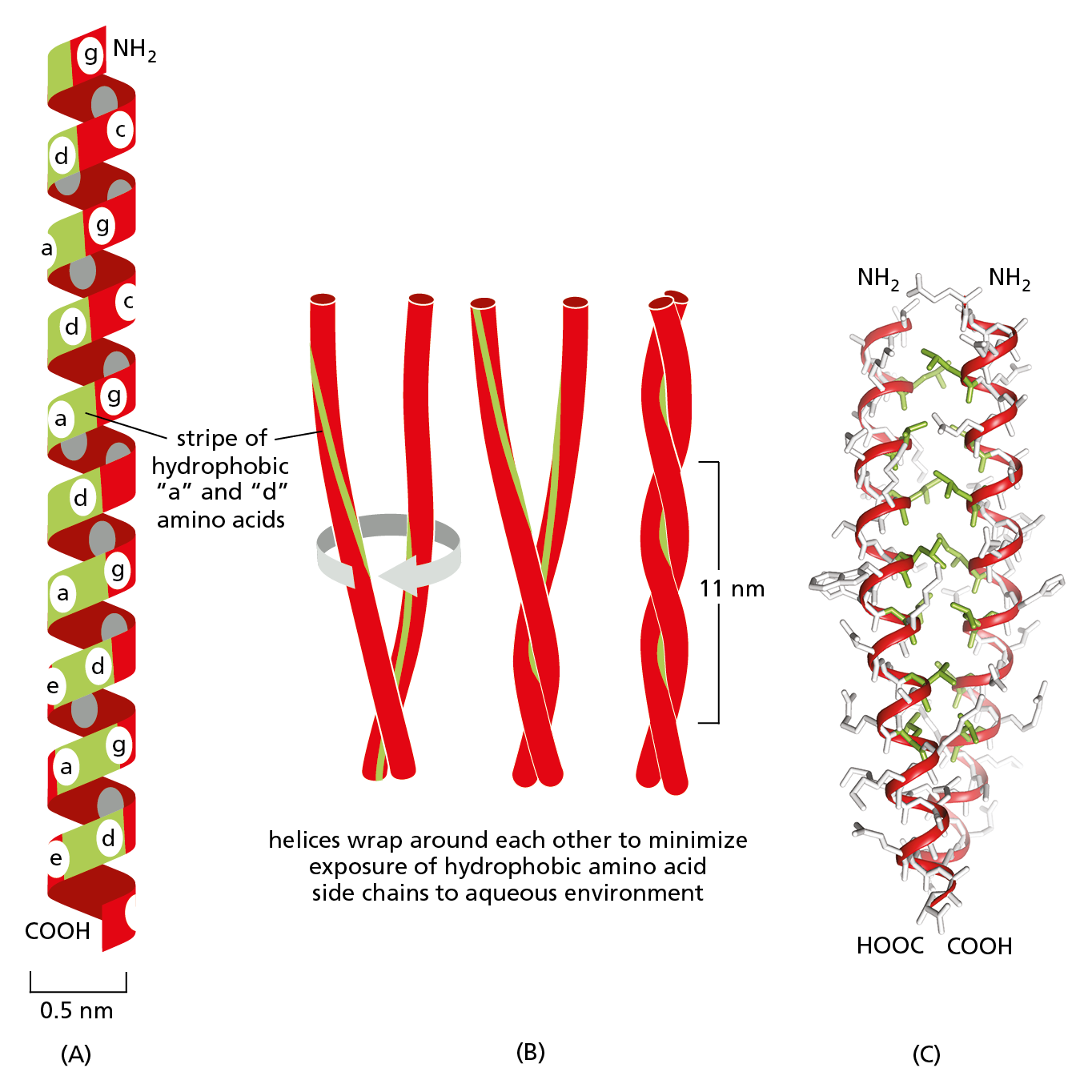
Figure 4−16 Intertwined α helices can form a stiff coiled-coil. (A) A single α helix is shown, with successive amino acid side chains labeled in a sevenfold repeating sequence “abcdefg.” Amino acids “a” and “d” in such a sequence lie close together on the cylinder surface, forming a stripe (shaded in green) that winds slowly around the α helix. Proteins that form coiled-coils typically have nonpolar amino acids at positions “a” and “d.” Consequently, as shown in (B), two α helices can wrap around each other, with the nonpolar side chains of one α helix interacting with the nonpolar side chains of the other, while the more hydrophilic amino acid side chains (shaded in red) are left exposed to the aqueous environment. (C) A portion of the atomic structure of a coiled-coil made by two α helices, as determined by x-ray crystallography. In this structure, the backbones of the helices are shown in red, the interacting, nonpolar side chains are green, and the remaining side chains are light gray. Coiled-coils can also form from three α helices (Movie 4.3).
β Sheets Form Rigid Structures at the Core of Many Proteins
A β sheet is made when hydrogen bonds form between segments of a polypeptide chain that lie side by side (see Figure 4−13A). When the neighboring segments run in the same orientation (say, from the N-terminus to the C-terminus), the structure forms a parallel β sheet; when they run in opposite directions, the structure forms an antiparallel β sheet (Figure 4−17). Both types of β sheet produce a very rigid, pleated structure, and they form the core of many proteins. Even the small bacterial transport protein HPr (see Figure 4−11) contains several β sheets.
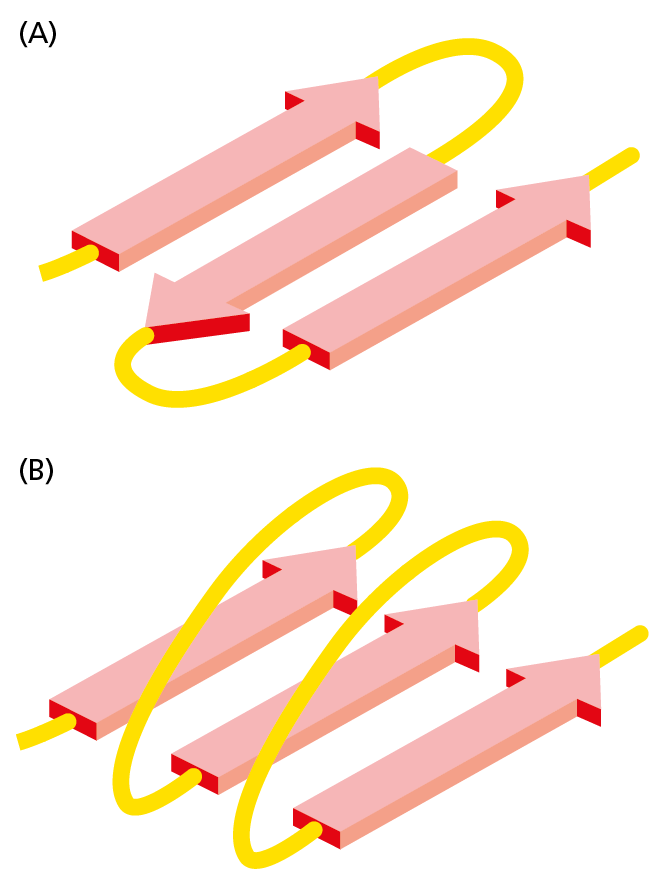
Figure 4−17 β sheets come in two varieties. (A) Antiparallel β sheet (see also Figure 4−13A). (B) Parallel β sheet. Both of these structures are common in proteins. By convention, the arrows point toward the C-terminus of the polypeptide chain (Movie 4.4).
β sheets have remarkable properties. They give silk fibers their extraordinary tensile strength. They also form the basis of amyloid structures, in which β sheets are stacked together in long rows with their amino acid side chains interdigitated like the teeth of a zipper (Figure 4−18). Such structures play an important role in cells, as we discuss later in this chapter. However, they can also precipitate disease, as we see next.
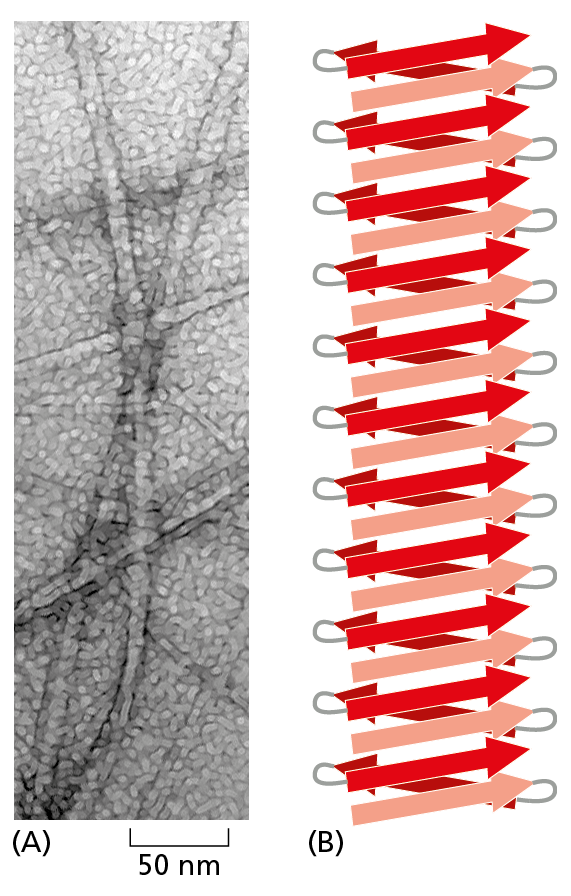
Figure 4−18 β sheets can stack to form an amyloid structure. (A) Electron micrograph showing an amyloid structure from a yeast. This structure resembles the type of insoluble aggregates observed in the neurons of individuals with different neurodegenerative diseases (see Figure 4−19). (B) Schematic representation shows the stacking of β sheets that stabilizes an individual amyloid strand. (A, from M.R. Sawaya et al., Nature 447:453–457, 2007. With permission from Macmillan Publishers Ltd.)
Misfolded Proteins Can Form Amyloid Structures That Cause Disease
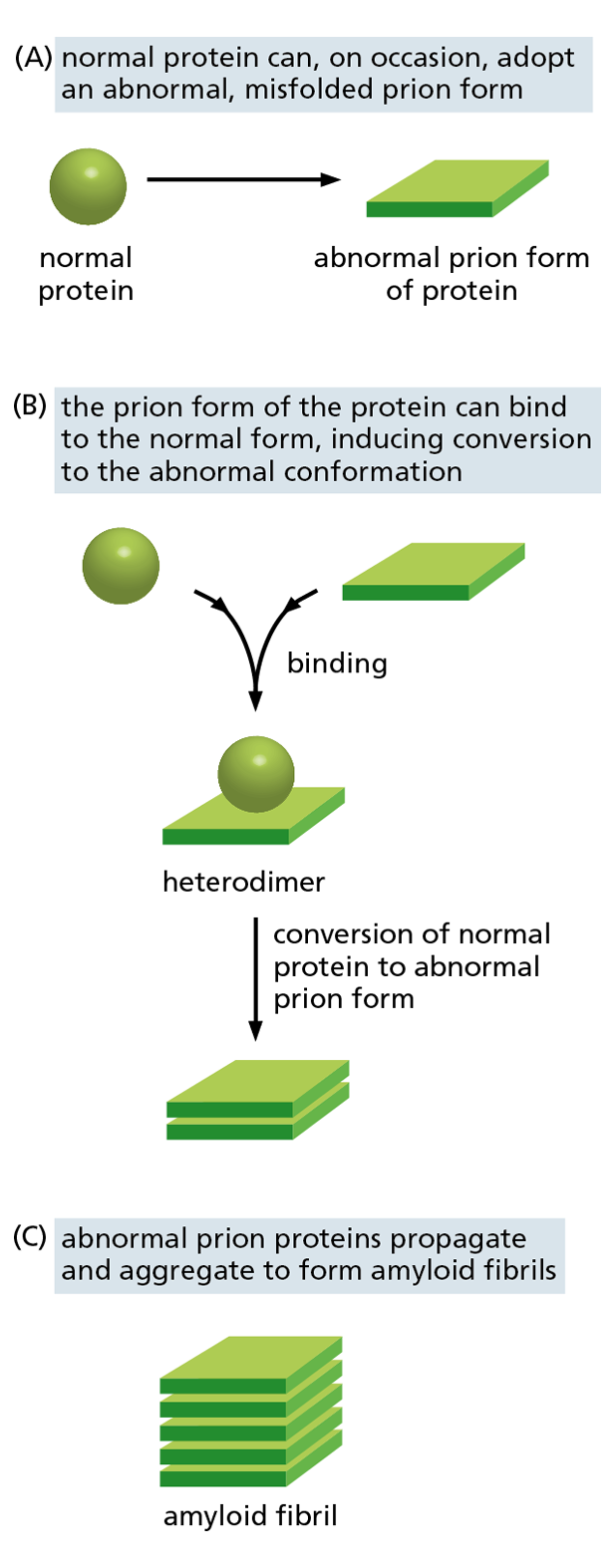
Figure 4−19 Prion diseases are caused by proteins whose misfolding is infectious. (A) A protein undergoes a rare conformational change to produce an abnormally folded prion form. (B) The abnormal form causes the conversion of normal proteins in the host’s brain into the misfolded prion form. (C) The prions aggregate into amyloid fibrils, which can disrupt brain-cell function, causing a neurodegenerative disorder (see also Figure 4–18). Some of the abnormal amyloid fibrils that form in major neurodegenerative disorders such as Alzheimer’s disease may be able to propagate from cell to cell in this way.
When proteins fold incorrectly, they sometimes form amyloid structures that can damage cells and even whole tissues. These amyloid struc-tures are thought to contribute to a number of neurodegenerative disorders, such as Alzheimer’s disease and Huntington’s disease. Some infectious neurodegenerative diseases—including scrapie in sheep, bovine spongiform encephalopathy (BSE, or “mad cow” disease) in cattle, and Creutzfeldt–Jakob disease (CJD) in humans—are caused by misfolded proteins called prions. The misfolded prion form of a protein can convert the properly folded version of the protein in an infected brain into the abnormal conformation. This allows the misfolded prions to form aggregates (Figure 4−19), which can spread rapidly from cell to cell, eventually causing the death of the affected animal or human. Prions are considered “infectious” because they can also spread from an affected individual to a normal individual via contaminated food, blood, or surgical instruments, for example.
Proteins Have Several Levels of Organization
A protein’s structure does not begin and end with α helices and β sheets. Its complete conformation includes several interdependent levels of organization, which build one upon the next. Because a protein’s structure begins with its amino acid sequence, this is considered its primary structure. The next level of organization includes the α helices and β sheets that form within certain segments of the polypeptide chain; these folds are elements of the protein’s secondary structure. The full, three-dimensional conformation formed by an entire polypeptide chain—including the α helices, β sheets, and all other loops and folds that form between the N- and C-termini—is sometimes referred to as the tertiary structure. Finally, if the protein molecule exists as a complex of more than one polypeptide chain, then these interacting polypeptides form its quaternary structure.
Studies of the conformation, function, and evolution of proteins have also revealed the importance of a level of organization distinct from the four just described. This organizational unit is the protein domain, which is defined as any segment of a polypeptide chain that can fold independently into a compact, stable structure. A protein domain usually contains between 40 and 350 amino acids—folded into α helices and β sheets and other elements of structure—and it is the modular unit from which many larger proteins are constructed (Figure 4−20).
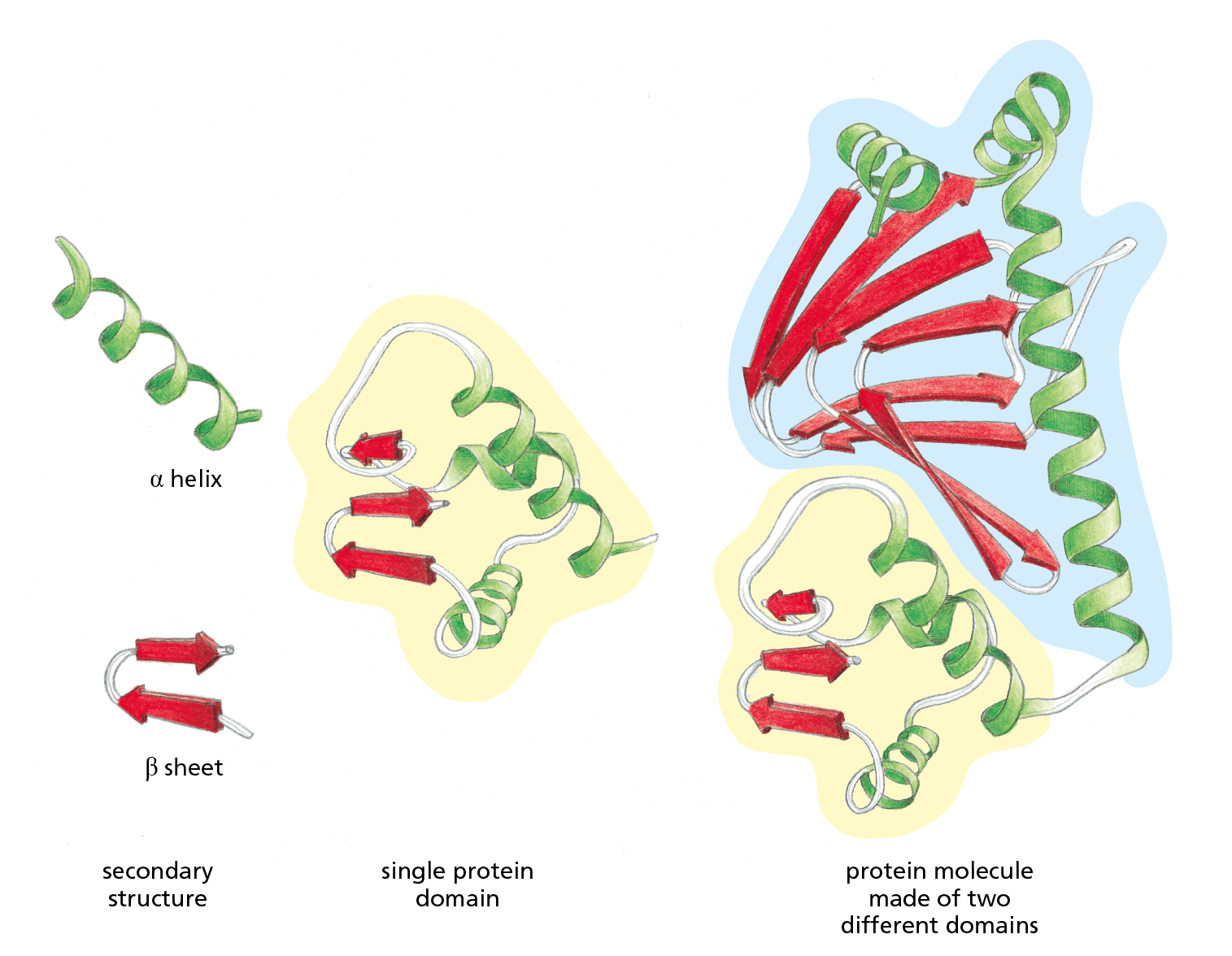
Figure 4−20 Many proteins are composed of separate functional domains. Elements of secondary structure such as α helices and β sheets pack together into stable, independently folding, globular elements called protein domains. A typical protein molecule is built from one or more domains, linked by a region of polypeptide chain that is often relatively unstructured. The ribbon diagram on the right represents the bacterial transcription regulatory protein CAP, which consists of one large cyclic AMP-binding domain (outlined in blue) and one small DNA-binding domain (outlined in yellow). The function of this protein is described in Chapter 8 (see Figure 8−9).
Different domains of a protein are often associated with different functions. For example, the bacterial catabolite activator protein (CAP), illustrated in Figure 4−20, has two domains: a small domain that binds to DNA and a large domain that binds cyclic AMP, a small intracellular signaling molecule. When the large domain binds cyclic AMP, it causes a conformational change in the protein that enables the small domain to bind to a specific DNA sequence and thereby promote the expression of an adjacent gene. To provide a sense of the many different domain structures observed in proteins, ribbon models of three different domains are shown in Figure 4−21.
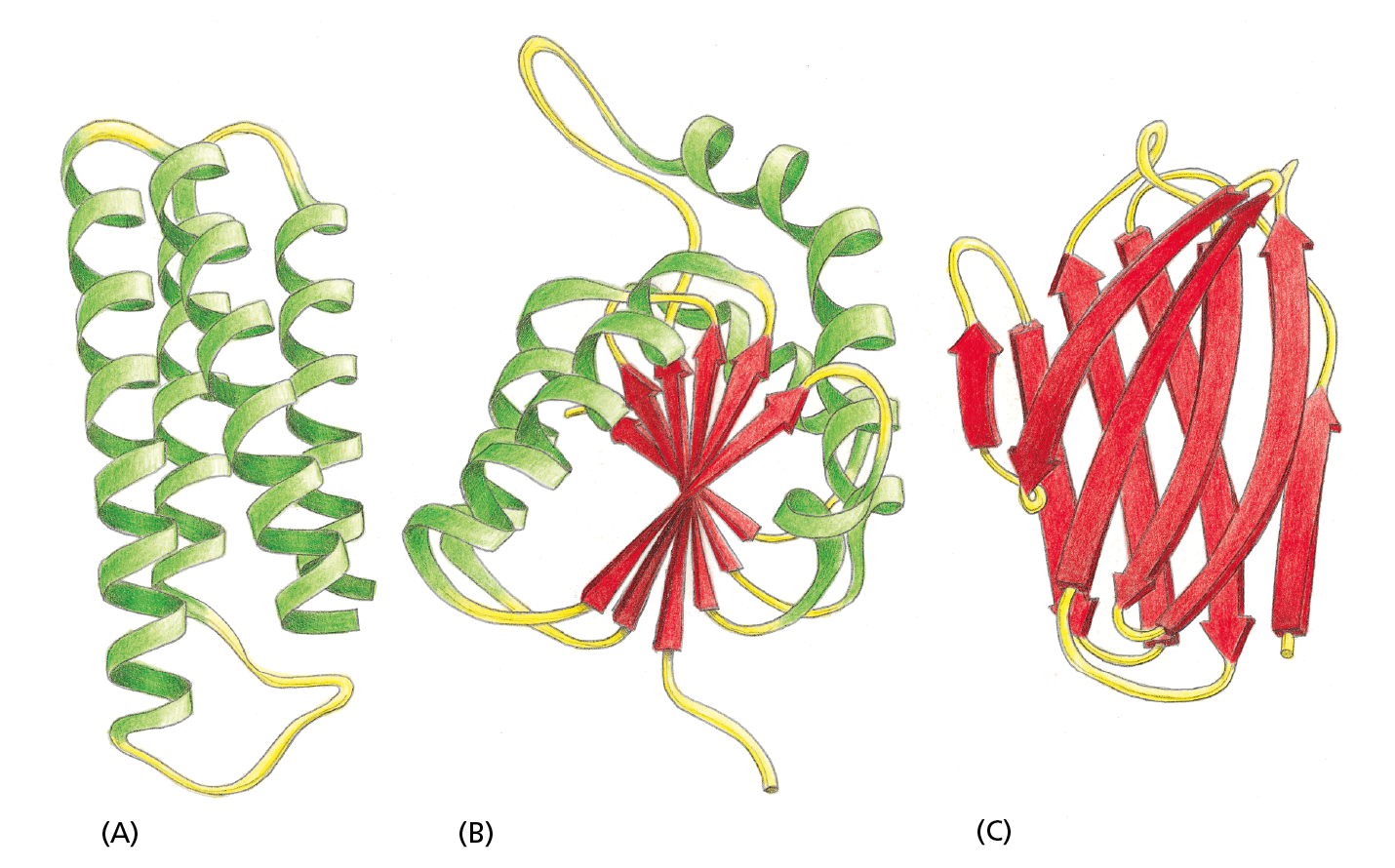
Figure 4−21 Ribbon models show three different protein domains. (A) Cytochrome b562 is a single-domain protein involved in electron transfer in E. coli. It is composed almost entirely of α helices. (B) The NAD-binding domain of the enzyme lactate dehydrogenase is composed of a mixture of α helices and β sheets. (C) An immunoglobulin domain of an antibody molecule is composed of a sandwich of two antiparallel β sheets. In these examples, the α helices are shown in green, while strands organized as β sheets are red. The protruding loop regions (yellow) are often unstructured and can provide binding sites for other molecules.
Proteins Also Contain Unstructured Regions
Small protein molecules, such as the oxygen-carrying muscle protein myoglobin, contain only a single domain (see Figure 4−10). Larger proteins can contain as many as several dozen domains, which are often connected by relatively short, unstructured lengths of polypeptide chain. The ubiquity of such intrinsically disordered sequences, which continually bend and flex due to thermal buffeting, became appreciated only after bioinformatics methods were developed that could recognize them from their amino acid sequences. Present estimates suggest that a third of all eukaryotic proteins also possess longer, unstructured regions—greater than 30 amino acids in length—in their polypeptide chains. These unstructured sequences can have a variety of important functions in cells, as we discuss later in the chapter.
Few of the Many Possible Polypeptide Chains Will Be Useful
In theory, a vast number of different polypeptide chains could be made from 20 different amino acids. Because each amino acid is chemically distinct and could, in principle, occur at any position, a polypeptide chain four amino acids long has 20 × 20 × 20 × 20 = 160,000 different possible sequences. For a typical protein with a length of 300 amino acids, that means that more than 20300 (that’s 10390) different polypeptide chains could theoretically be produced. And that’s just one protein.
Of the unimaginably large collection of potential polypeptide sequences, only a minuscule fraction is actually made by cells. That’s because most biological functions depend on proteins with stable, well-defined three-dimensional conformations. This requirement greatly restricts the list of polypeptide sequences present in living cells. Another constraint is that functional proteins must be “well-behaved” and not engage in unwanted associations with other proteins in the cell—forming insoluble protein aggregates, for example. Many potential protein sequences would therefore have been eliminated by natural selection through the long trial-and-error process that underlies evolution (discussed in Chapter 9).
Thanks to natural selection, the amino acid sequences of many present-day polypeptides have evolved to adopt a stable conformation—one that bestows upon the protein the exact chemical properties that will enable it to perform a particular function. Such proteins are so precisely built that a change in even a few atoms in one amino acid can sometimes disrupt the structure of a protein and thereby eliminate its function. In fact, the conformations of many proteins—and their constituent domains—are so stable and effective that they have been conserved throughout the evolution of a diverse array of organisms. For example, the three-dimensional structures of the DNA-binding domains of some transcription regulators from yeast, animals, and plants are almost completely superimposable, even though the organisms are separated by more than a billion years of evolution. Other proteins, however, have changed their structure and function over evolutionary time, as we now discuss.
Proteins Can Be Classified into Families
Once a protein has evolved a stable conformation with useful properties, its structure can be modified over time to enable it to perform new functions. We know that this occurred quite often during evolution, because many present-day proteins can be grouped into protein families, in which each family member has an amino acid sequence and a three-dimensional conformation that closely resemble those of the other family members.
Question 4–3
Random mutations only very rarely result in changes that improve a protein’s usefulness for the cell, yet useful mutations are selected in evolution. Because these changes are so rare, for each useful mutation there are innumerable mutations that lead to either no improvement or inactive proteins. Why, then, do cells not contain millions of proteins that are of no use?
Consider, for example, the serine proteases, a family of protein-cleaving (proteolytic) enzymes that includes the digestive enzymes chymotrypsin, trypsin, and elastase, as well as several proteases involved in blood clotting. When any two of these enzymes are compared, portions of their amino acid sequences are found to be nearly the same. The similarity of their three-dimensional conformations is even more striking: most of the detailed twists and turns in their polypeptide chains, which are several hundred amino acids long, are virtually identical (Figure 4−22). The various serine proteases nevertheless have distinct enzymatic activities, each cleaving different proteins or the peptide bonds between different types of amino acids.
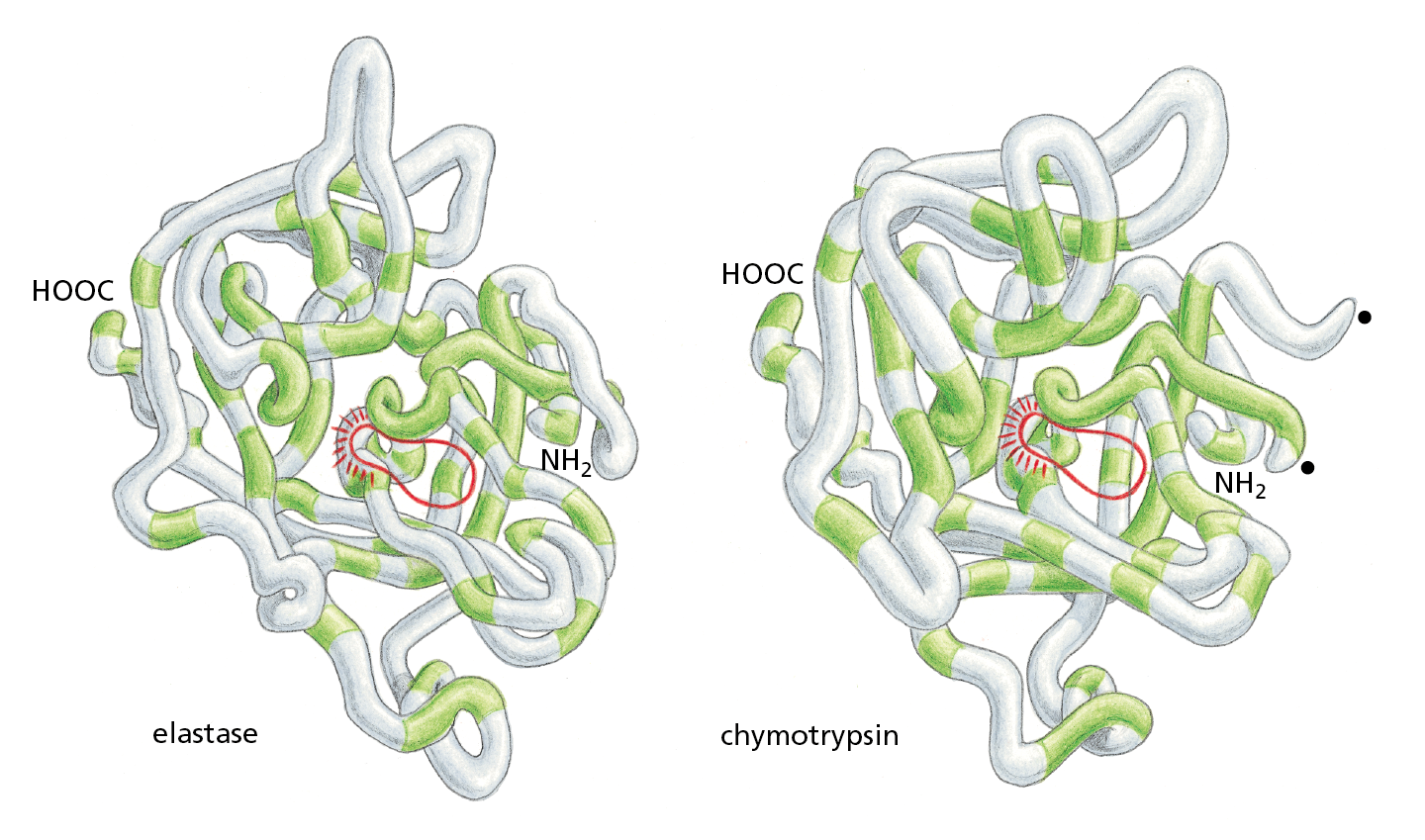
Figure 4−22 Serine proteases constitute a family of proteolytic enzymes. Backbone models of two serine proteases, elastase and chymotrypsin, are illustrated. Although only those amino acid sequences in the polypeptide chain shaded in green are the same in the two proteins, the two conformations are very similar nearly everywhere. Nonetheless, the two proteases act on different substrates.
The active site of each enzyme—where its substrates are bound and cleaved—is circled in red. The amino acid serine directly participates in the cleavage reaction, which is why the enzymes are called serine proteases. The black dots on the right side of the chymotrypsin molecule mark the two ends created where the enzyme has cleaved its own backbone.
Large Protein Molecules Often Contain More than One Polypeptide Chain
The same type of weak noncovalent bonds that enable a polypeptide chain to fold into a specific conformation also allow proteins to bind to each other to produce larger structures in the cell. Any region on a protein’s surface that interacts with another molecule through sets of noncovalent bonds is termed a binding site. A protein can contain binding sites for a variety of molecules, large and small. If a binding site recognizes the surface of a second protein, the tight binding of two folded polypeptide chains at this site will create a larger protein, whose quaternary structure has a precisely defined geometry. Each polypeptide chain in such a protein is called a subunit, and each of these subunits may contain more than one domain.
In the simplest case, two identical, folded polypeptide chains form a symmetrical complex of two protein subunits (called a dimer) that is held together by interactions between two identical binding sites. CAP, the bacterial protein we discussed earlier, is such a dimer (Figure 4−23A); it is composed of two identical copies of the protein subunit, each of which contains two domains, as shown previously in Figure 4−20. Many other symmetrical protein complexes, formed from multiple copies of the same polypeptide chain, are commonly found in cells. The enzyme neuraminidase, for example, consists of a ring of four identical protein subunits (Figure 4−23B).
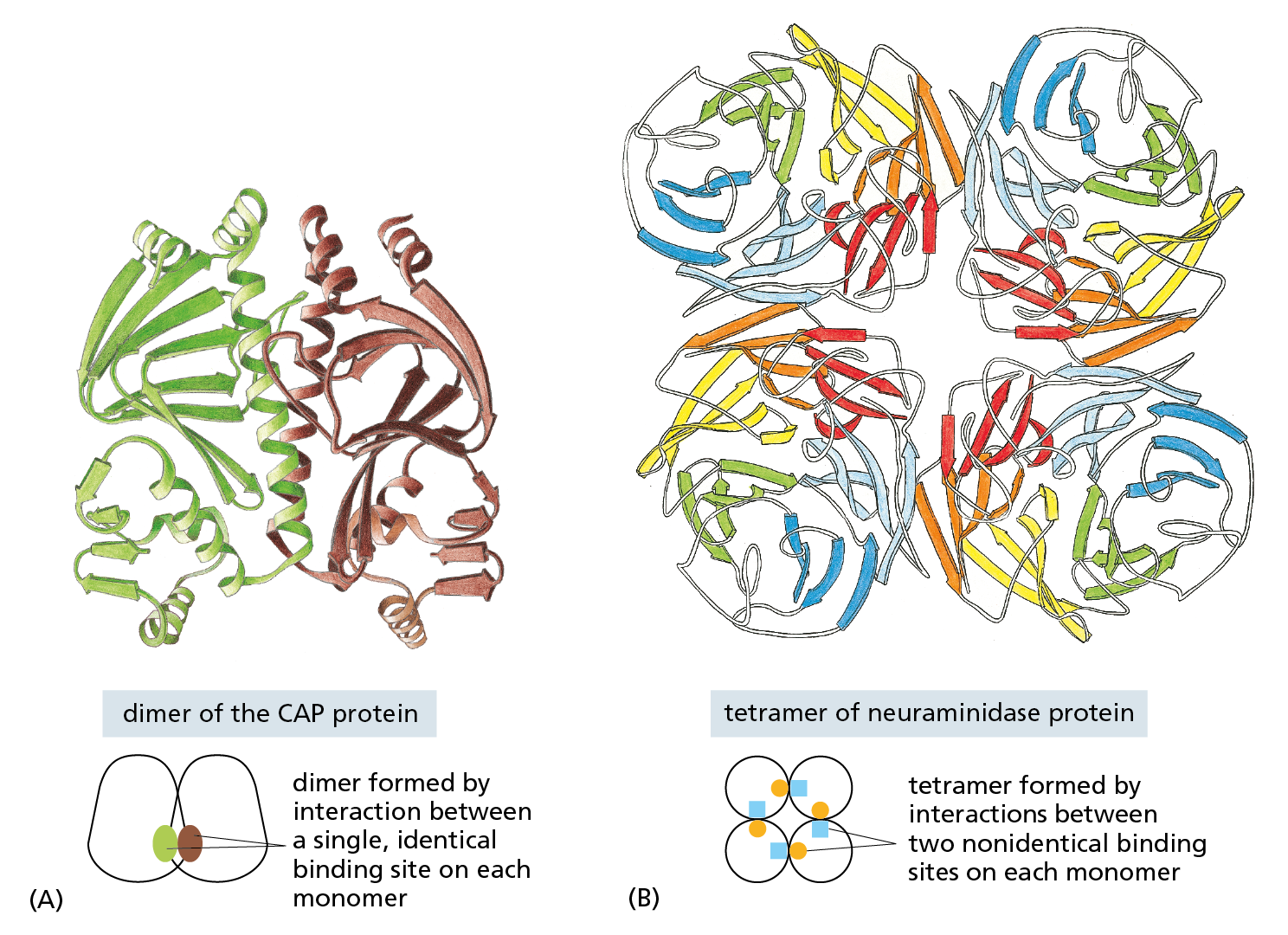
Figure 4−23 Many protein molecules contain multiple copies of the same protein subunit. (A) A symmetrical dimer. The protein CAP is a complex of two identical polypeptide chains (see also Figure 4–20). (B) A symmetrical homotetramer. The enzyme neuraminidase exists as a ring of four identical polypeptide chains. For both (A) and (B), a small schematic below the structure emphasizes how the repeated use of the same binding interaction forms the structure. In (A), the use of the same binding site on each monomer (represented by brown and green ovals) causes the formation of a symmetrical dimer. In (B), a pair of nonidentical binding sites (represented by orange circles and blue squares) causes the formation of a symmetrical tetramer.
Other proteins contain two or more different polypeptide chains. Hemoglobin, the protein that carries oxygen in red blood cells, is a particularly well-studied example. The protein contains two identical α-globin subunits and two identical β-globin subunits, symmetrically arranged (Figure 4−24). Many proteins contain multiple subunits, and they can be very large (Movie 4.5).
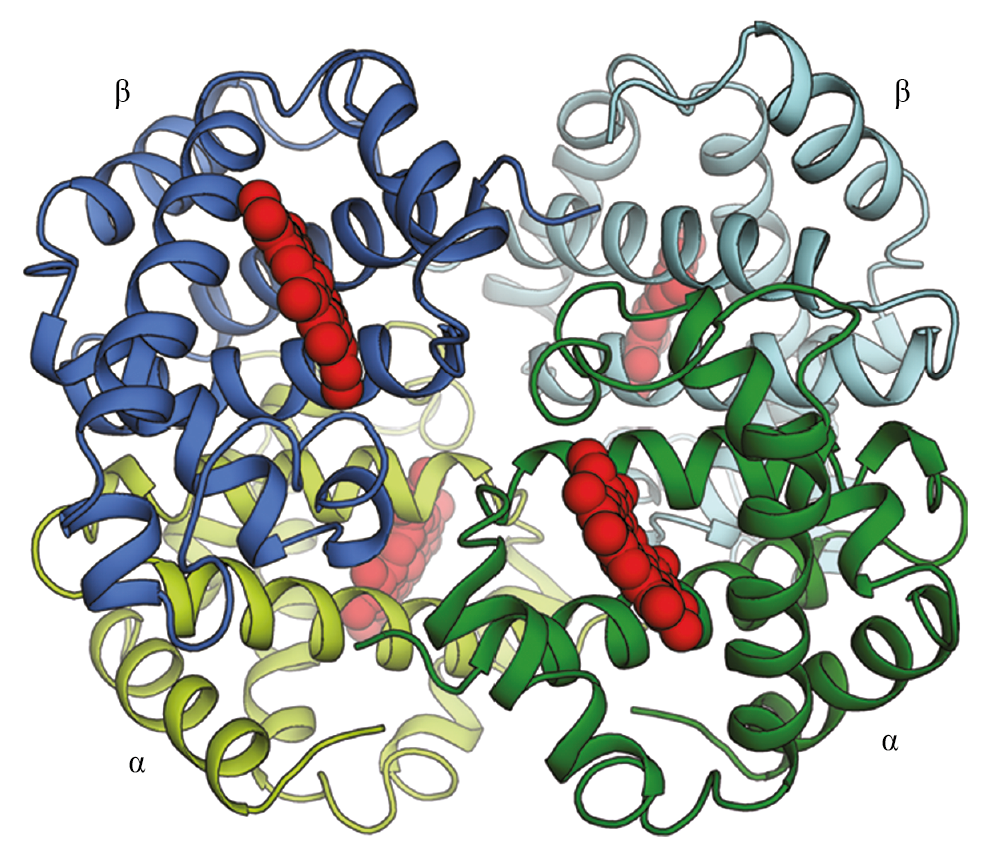
Figure 4−24 Some proteins are formed as a symmetrical assembly of two different subunits. Hemoglobin, an oxygen-carrying protein abundant in red blood cells, contains two copies of α-globin (green) and two copies of β-globin (blue). Each of these four polypeptide chains cradles a molecule of heme (red), where oxygen (O2) is bound. Thus, each hemoglobin protein can carry four molecules of oxygen.
Proteins Can Assemble into Filaments, Sheets, or Spheres
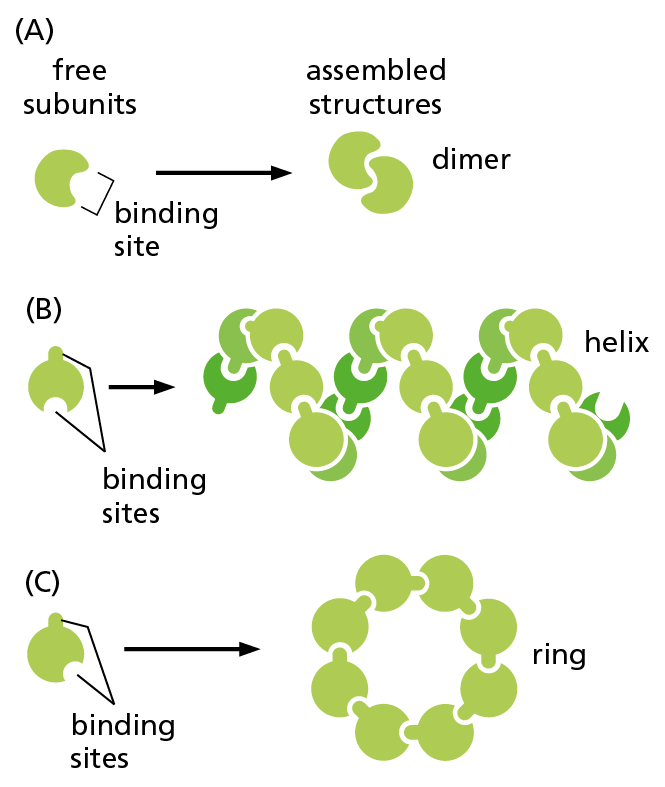
Figure 4−25 Identical protein subunits can assemble into complex structures. (A) A protein with just one binding site can form a dimer with another identical protein. (B) Identical proteins with two different binding sites will often form a long, helical filament. (C) If the two binding sites are positioned appropriately in relation to each other, the protein subunits will form a closed ring instead of a helix (see also Figure 4−23B).
Proteins can form even larger assemblies than those discussed so far. Most simply, a chain of identical protein molecules can be formed if the binding site on one protein molecule is complementary to another region on the surface of another protein molecule of the same type. Because each protein molecule is bound to its neighbor in an identical way (see Figure 4−14), the molecules will often be arranged in a helix that can be extended indefinitely in either direction (Figure 4−25). This type of arrangement can produce an extended protein filament. An actin filament, for example, is a long, helical structure formed from many molecules of the protein actin (Figure 4−26). Actin is extremely abundant in eukaryotic cells, where it forms one of the major filament systems of the cytoskeleton (discussed in Chapter 17). Other sets of identical proteins associate to form tubes, as in the microtubules of the cytoskeleton (Figure 4−27), or cagelike spherical shells, as in the protein coats of virus particles (Figure 4−28).
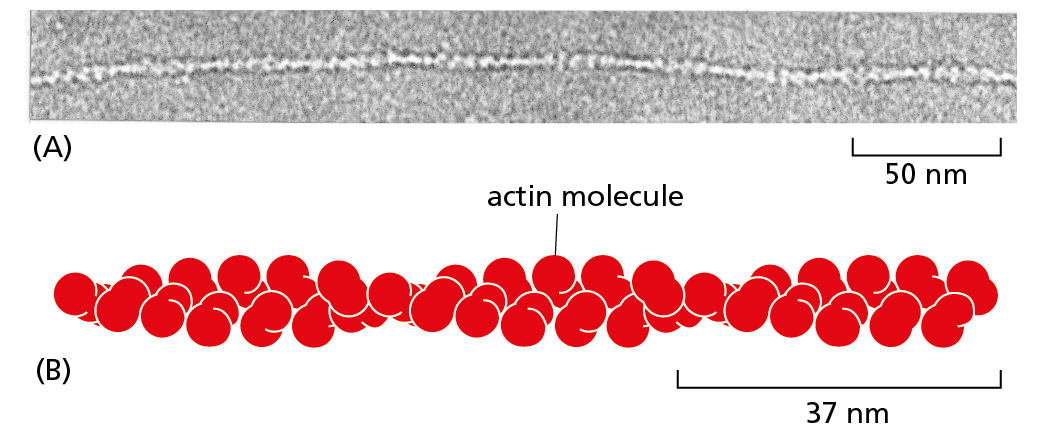
Figure 4–26 An actin filament is composed of identical protein subunits. (A) Transmission electron micrograph of an actin filament. (B) The helical array of actin molecules in an actin filament often contains thousands of molecules and extends for micrometers in the cell; 1 micrometer = 1000 nanometers. (A, courtesy of Roger Craig.)
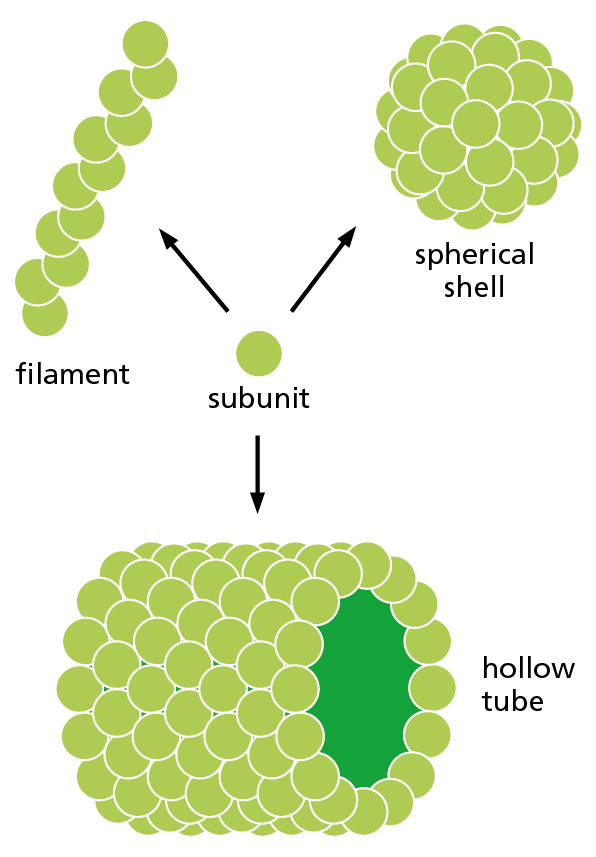
Figure 4−27 A single type of protein subunit can pack together to form a filament, a hollow tube, or a spherical shell. Actin subunits, for example, form actin filaments (see Figure 4–26), whereas tubulin subunits form hollow microtubules, and some virus proteins form a spherical shell (capsid) that encloses the viral genome (see Figure 4−28).
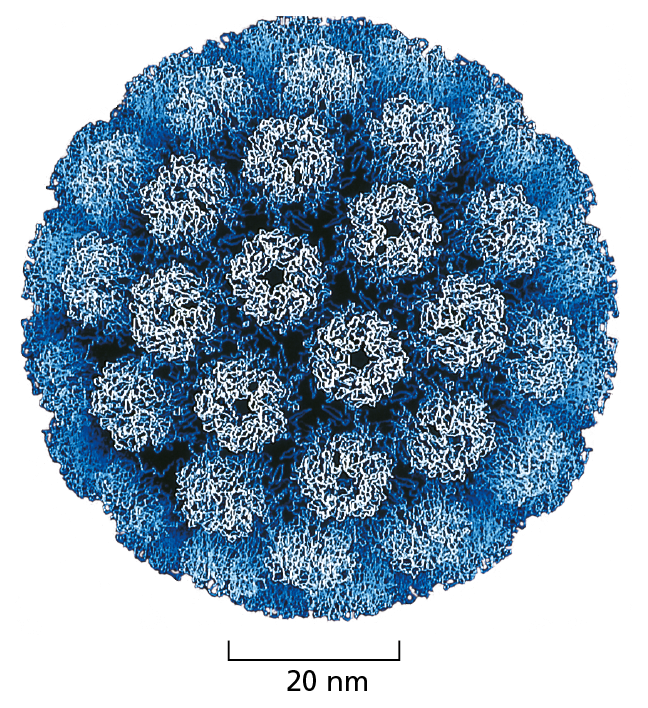
Figure 4−28 Many viral capsids are essentially spherical protein assemblies. They are formed from many copies of a small set of protein subunits. The nucleic acid of the virus (DNA or RNA) is packaged inside. The structure of the simian virus SV40, shown here, was determined by x-ray crystallography and is known in atomic detail. (Courtesy of Robert Grant, Stephan Crainic, and James M. Hogle.)
Many large structures, such as viruses and ribosomes, are built from a mixture of one or more types of protein plus RNA or DNA molecules. These structures can be isolated in pure form and dissociated into their constituent macromolecules. It is often possible to mix the isolated components back together and watch them reassemble spontaneously into the original structure. This demonstrates that all the information needed for assembly of the complicated structure is contained in the macromolecules themselves. Experiments of this type show that much of the structure of a cell is self-organizing: if the required proteins are produced in the right amounts, the appropriate structures will form automatically.
Some Types of Proteins Have Elongated Fibrous Shapes
Most of the proteins we have discussed so far are globular proteins, in which the polypeptide chain folds up into a compact shape like a ball with an irregular surface. Enzymes, for example, tend to be globular proteins: even though many are large and complicated, with multiple subunits, most have a quaternary structure with an overall rounded shape (see Figure 4−10). In contrast, other proteins have roles in the cell that require them to span a large distance. These proteins generally have a relatively simple, elongated three-dimensional structure and are commonly referred to as fibrous proteins.
One large class of intracellular fibrous proteins resembles α-keratin, which we met earlier when we introduced the α helix. Keratin filaments are extremely stable: long-lived structures such as hair, horns, and nails are composed mainly of this protein. An α-keratin molecule is a dimer of two identical subunits, with the long α helices of each subunit forming a coiled-coil (see Figure 4−16). These coiled-coil regions are capped at either end by globular domains containing binding sites that allow them to assemble into ropelike intermediate filaments—a component of the cytoskeleton that gives cells mechanical strength (discussed in Chapter 17).
Fibrous proteins are especially abundant outside the cell, where they form the gel-like extracellular matrix that helps bind cells together to form tissues. These proteins are secreted by the cells into their surroundings, where they often assemble into sheets or long fibrils. Collagen is the most abundant of these fibrous extracellular proteins in animal tissues. A collagen molecule consists of three long polypeptide chains, each containing the nonpolar amino acid glycine at every third position. This regular structure allows the chains to wind around one another to generate a long, regular, triple helix with glycine at its core (Figure 4−29A). Many such collagen molecules bind to one another, side-by-side and end-to-end, to create long, overlapping arrays called collagen fibrils, which are extremely strong and help hold tissues together, as described in Chapter 20.
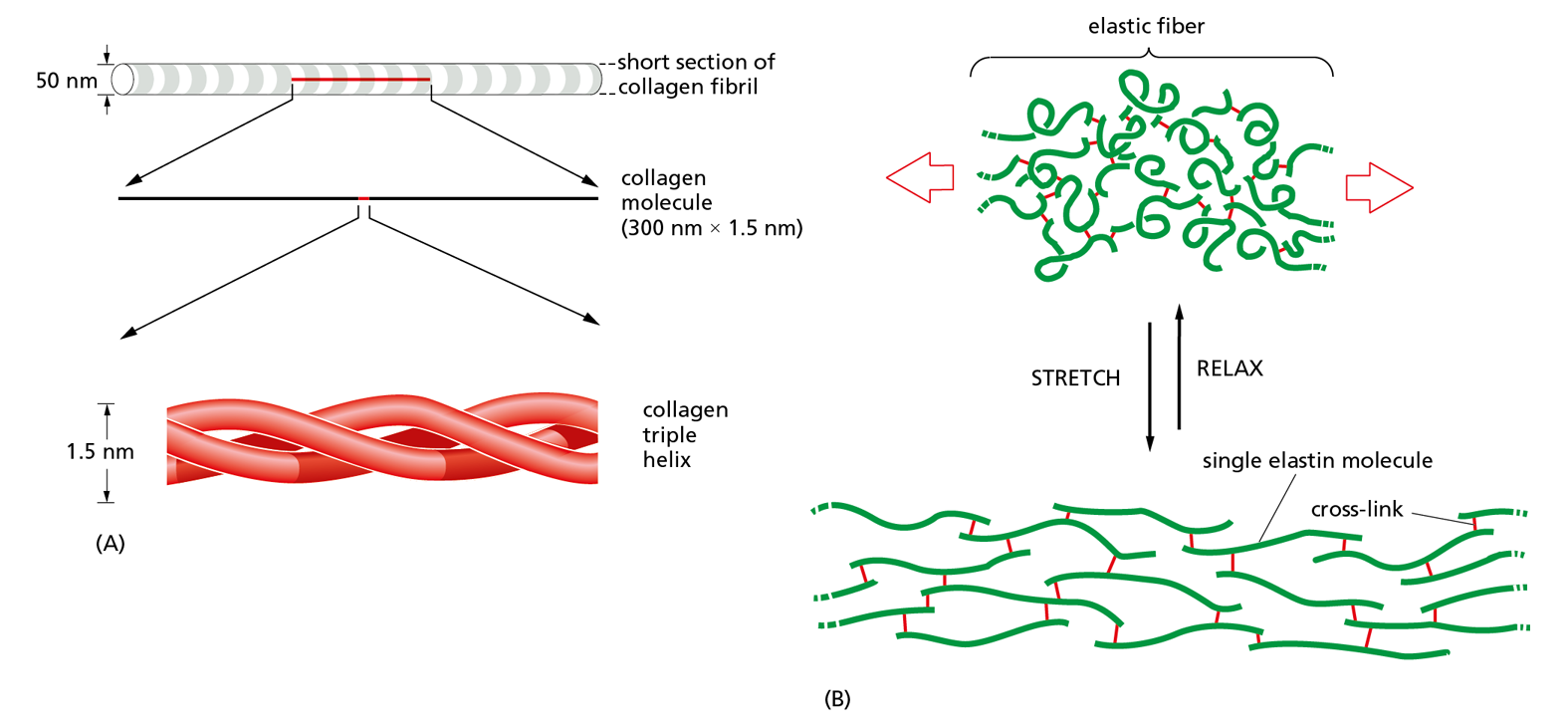
Figure 4−29 Fibrous proteins collagen and elastin form very different structures. (A) A collagen molecule is a triple helix formed by three extended protein chains that wrap around one another. Many rodlike collagen molecules are cross-linked together in the extracellular space to form collagen fibrils (top), which have the tensile strength of steel. The striping on the collagen fibril is caused by the regular repeating arrangement of the collagen molecules within the fibril. (B) Elastin molecules are cross-linked together by covalent bonds (red) to form rubberlike, elastic fibers. Each elastin polypeptide chain uncoils into a more extended conformation when the fiber is stretched, and recoils spontaneously as soon as the stretching force is relaxed.
In complete contrast to collagen is another fibrous protein in the extracellular matrix, elastin. Elastin molecules are formed from relatively loose and unstructured polypeptide chains that are covalently cross-linked into a rubberlike elastic meshwork. The resulting elastic fibers enable skin and other tissues, such as arteries and lungs, to stretch and recoil without tearing. As illustrated in Figure 4−29B, the elasticity is due to the ability of the individual protein molecules to uncoil reversibly whenever they are stretched.
Extracellular Proteins Are Often Stabilized by Covalent Cross-Linkages
Many protein molecules are attached to the surface of a cell’s plasma membrane or secreted as part of the extracellular matrix, which exposes them to the potentially harsh conditions outside the cell. To help maintain their structures, the polypeptide chains in such proteins are often stabilized by covalent cross-linkages. These linkages can either tie together two amino acids in the same polypeptide chain or join together many polypeptide chains in a large protein complex—as for the collagen fibrils and elastic fibers just described. A variety of different types of cross-links exist.
The most common covalent cross-links in proteins are sulfur–sulfur bonds. These disulfide bonds (also called S–S bonds) are formed, before a protein is secreted, by an enzyme in the endoplasmic reticulum that links together two –SH groups from cysteine side chains that are adjacent in the folded protein (Figure 4−30). Disulfide bonds do not change a protein’s conformation, but instead act as a sort of “atomic staple” to reinforce the protein’s most favored conformation. Lysozyme—an enzyme in tears, saliva, and other secretions that can disrupt bacterial cell walls—retains its antibacterial activity for a long time because it is stabilized by such disulfide cross-links.
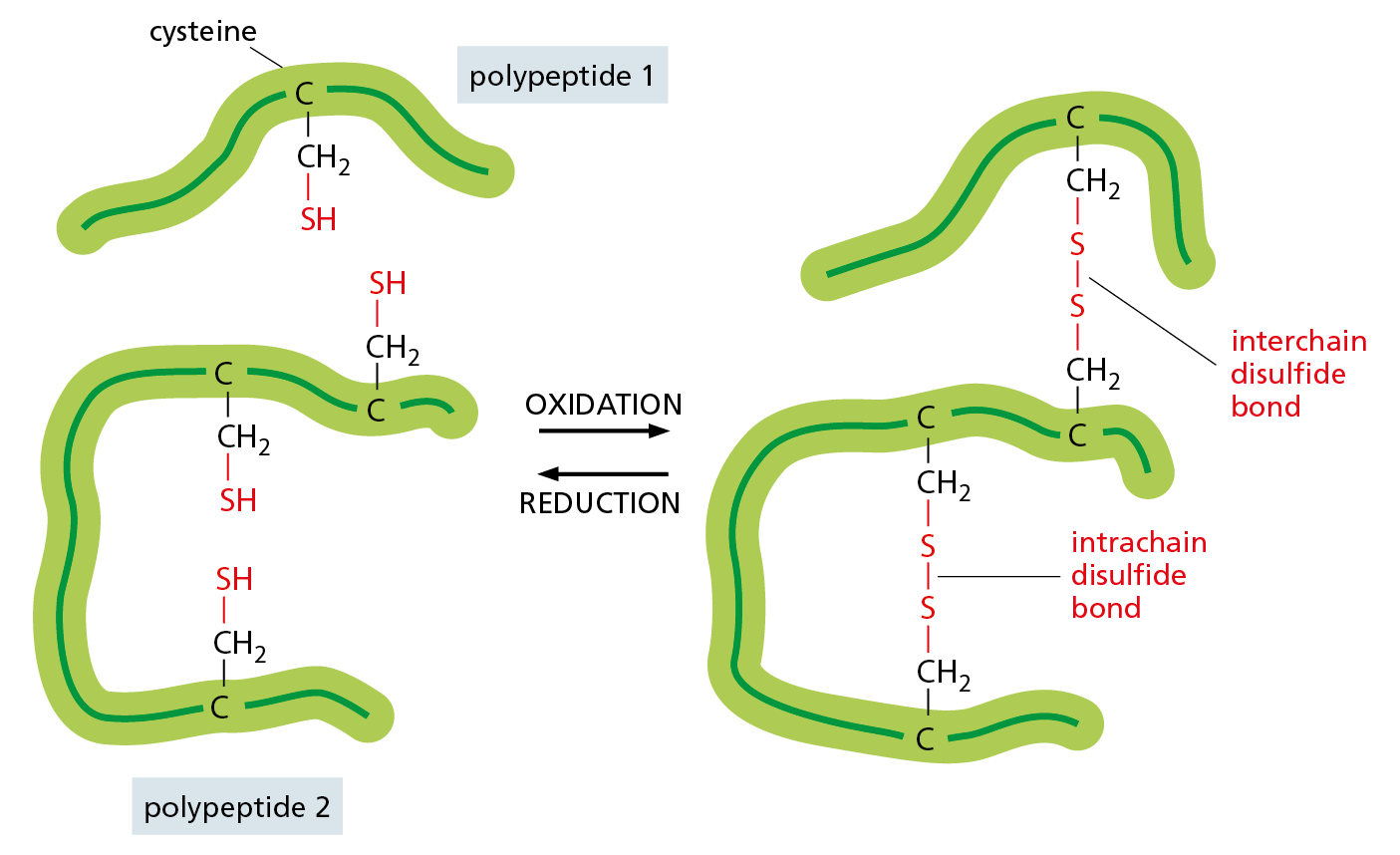
Figure 4−30 Disulfide bonds help stabilize a favored protein conformation. This diagram illustrates how covalent disulfide bonds form between adjacent cysteine side chains by the oxidation of their –SH groups. As indicated, these cross-links can join either two parts of the same polypeptide chain or two different polypeptide chains. Because the energy required to break one covalent bond is much larger than the energy required to break even a whole set of noncovalent bonds (see Table 2−1, p. 48), a disulfide bond can have a major stabilizing effect on a protein’s folded structure (Movie 4.6).
Disulfide bonds generally do not form in the cell cytosol, where a high concentration of reducing agents converts such bonds back to cysteine–SH groups. Apparently, proteins do not require this type of structural reinforcement in the relatively mild conditions inside the cell.
- peptide bond
Covalent chemical bond between the carbonyl group of one amino acid and the amino group of a second amino acid. (See Panel 2–6, pp. 76–77.)
- polypeptide, polypeptide chain
Linear polymer composed of multiple amino acids. Proteins are composed of one or more long polypeptide chains.
- amino acid sequence
The order of the amino acid subunits in a protein chain. Sometimes called the primary structure of a protein.
- polypeptide backbone
Repeating sequence of the atoms (–N–C–C–) that form the core of a protein molecule and to which the amino acid side chains are attached.
- N-terminus
The end of a polypeptide chain that carries a free α-amino group.
- C-terminus
The end of a polypeptide chain that carries a free carboxyl group (–COOH).
- side chain
Portion of an amino acid not involved in forming peptide bonds; its chemical identity gives each amino acid unique properties.
- conformation
Precise, three-dimensional shape of a protein or other macromolecule, based on the spatial location of its atoms in relation to one another.
- alpha helix
Folding pattern, common in many proteins, in which a single polypeptide chain twists around itself to form a rigid cylinder stabilized by hydrogen bonds between every fourth amino acid.
- beta sheet (βsheet)
Folding pattern found in many proteins in which neighboring regions of the polypeptide chain associate side-by-side with each other through hydrogen bonds to give a rigid, flattened structure.
- helix
An elongated structure whose subunits twist in a regular fashion around a central axis, like a spiral staircase.
- coiled-coil
Stable, rodlike protein structure formed when two or more α helices twist repeatedly around each other.
- primary structure
The amino acid sequence of a protein.
- secondary structure
Regular local folding pattern of a polymeric molecule. In proteins, it refers to α helices and β sheets.
- tertiary structure
Complete three-dimensional structure of a fully folded protein.
- quaternary structure
Complete structure formed by multiple, interacting polypeptide chains that form a larger protein molecule.
- protein domain
Segment of a polypeptide chain that can fold into a compact, stable structure and that often carries out a specific function.
- intrinsically disordered sequence
Region in a polypeptide chain that lacks a definite structure.
- protein family
A group of polypeptides that share a similar amino acid sequence or three-dimensional structure, reflecting a common evolutionary origin. Individual members often have related but distinct functions, such as kinases that phosphorylate different target proteins.
- subunit
A monomer that forms part of a larger molecule, such as an amino acid residue in a protein or a nucleotide residue in a nucleic acid. Can also refer to a complete molecule that forms part of a larger molecule. Many proteins, for example, are composed of multiple polypeptide chains, each of which is called a protein subunit.
- globular protein
Any protein in which the polypeptide chain folds into a compact, rounded shape. Includes most enzymes.
- fibrous protein
A protein with an elongated, rodlike shape, such as collagen or a keratin filament.
- disulfide bond
Covalent cross-link formed between the sulfhydryl groups on two cysteine side chains; often used to reinforce a secreted protein’s structure or to join two different proteins together.