DNA REPLICATION MECHANISMS
All organisms duplicate their DNA with extraordinary accuracy before each cell division. In this part of the chapter, we explore how an elaborate “replication machine” achieves this accuracy, while duplicating DNA at rates as high as 1000 nucleotides per second.
Base-pairing Underlies DNA Replication and DNA Repair
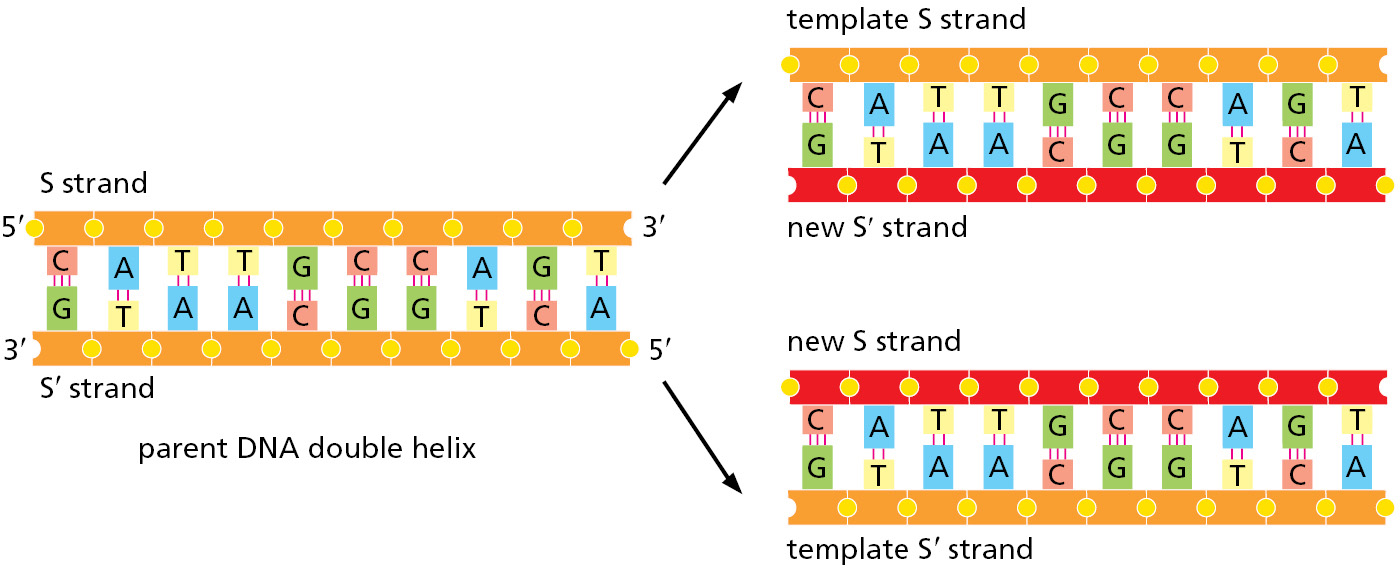
As introduced in Chapter 1, DNA templating is the mechanism the cell uses to copy the nucleotide sequence of one DNA strand into a complementary DNA sequence (Figure 5–2). This process requires the separation of the DNA helix into two template strands, and it entails the recognition of each nucleotide in the DNA template strands by a free (unpolymerized) complementary nucleotide. The separation of the DNA helix exposes the hydrogen-bond donor and acceptor groups on each DNA base to allow its base-pairing with the appropriate incoming free nucleotide, aligning it for its enzyme-catalyzed polymerization into a new DNA chain.
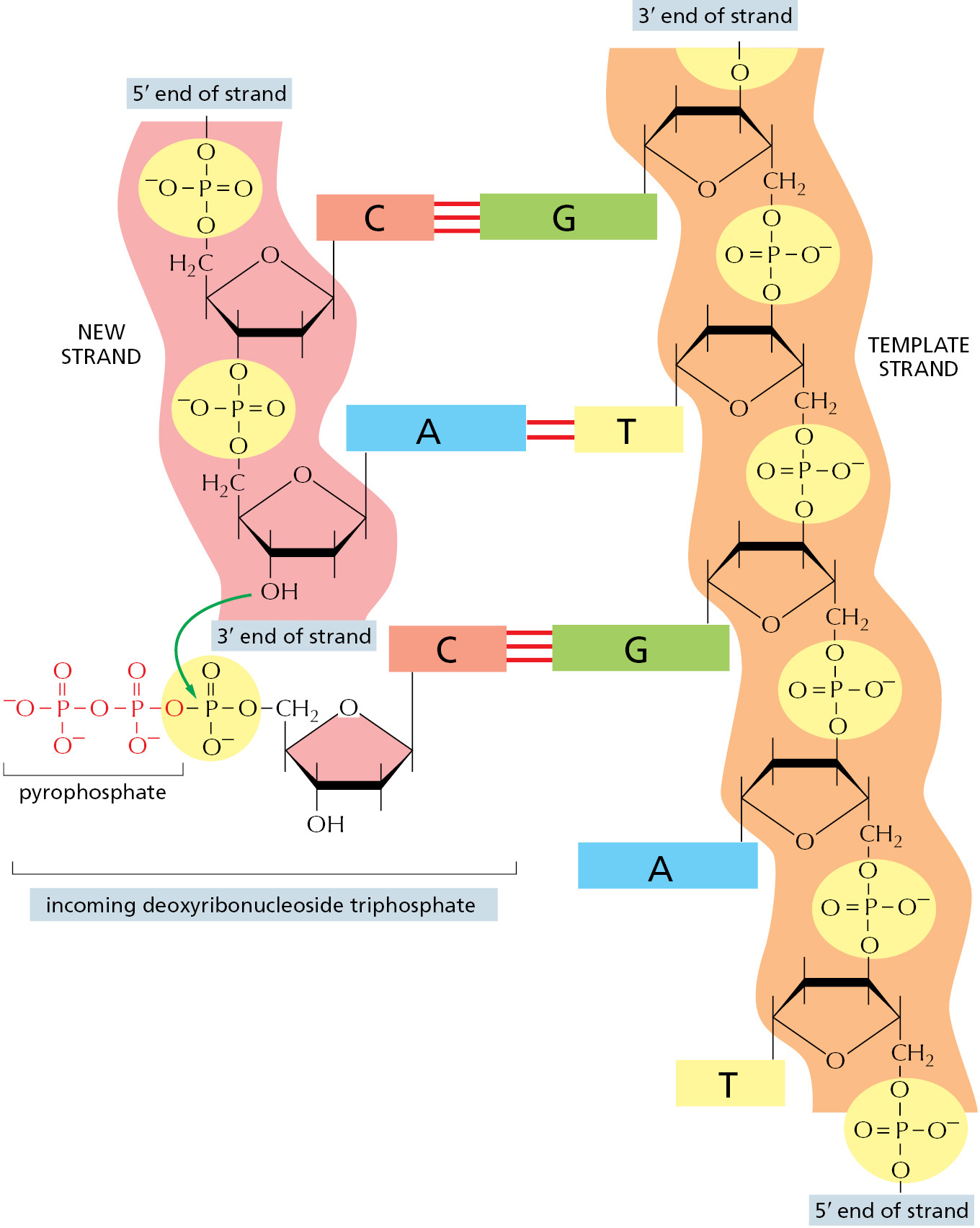
The first nucleotide-polymerizing enzyme, DNA polymerase, was discovered in 1957. The free nucleotides that serve as substrates for this enzyme were found to be deoxyribonucleoside triphosphates, and their polymerization into DNA required a single-strand DNA template. Figure 5–3 and Figure 5–4 illustrate the stepwise mechanism of this reaction.
{kind=link}
The DNA Replication Fork Is Asymmetrical
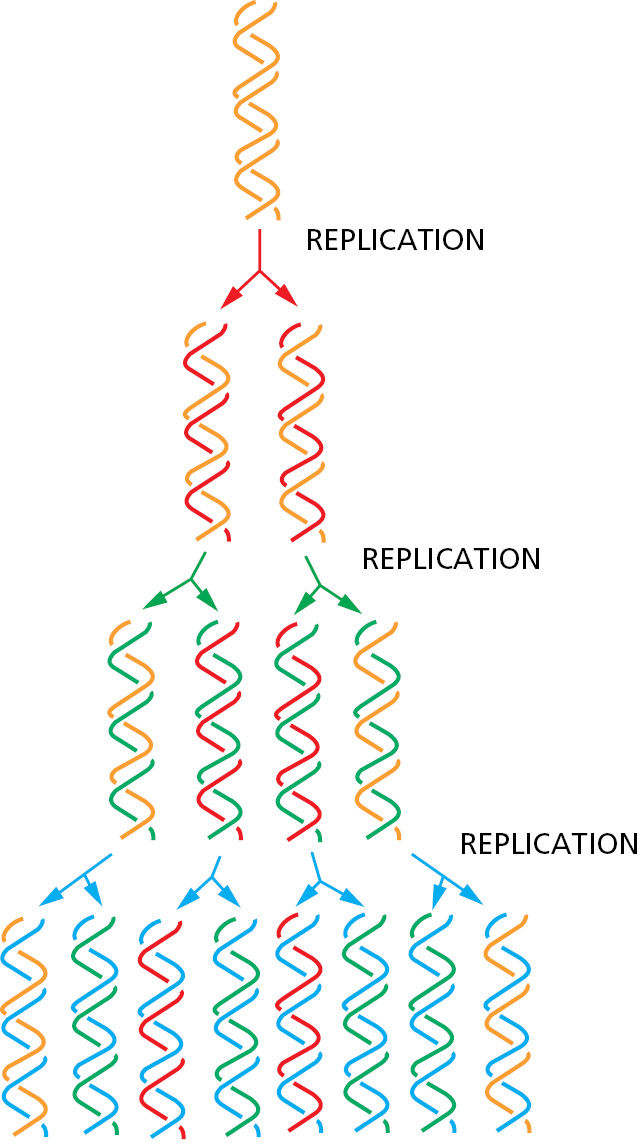
During DNA replication inside a cell, each of the two original DNA strands serves as a template for the formation of an entire new strand. Because each of the two daughters of a dividing cell inherits a new DNA double helix containing one original and one new strand (Figure 5–5), the DNA double helix is said to be replicated semiconservatively. How is this feat actually accomplished?
Analyses carried out in the early 1960s on the whole replicating chromosome of an E. coli bacterium revealed a localized region of replication that moves progressively along the parent DNA double helix. Because of its Y-shaped structure, this active region is called a replication fork (Figure 5–6). At the replication fork, a multienzyme complex that contains the DNA polymerase synthesizes the DNA of both new daughter strands.
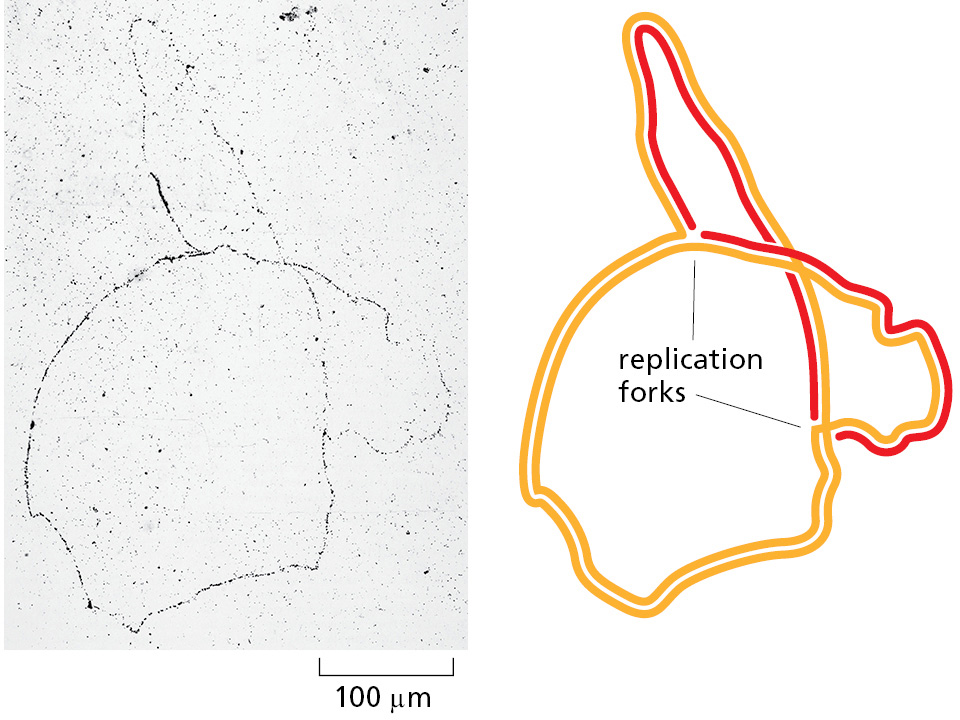
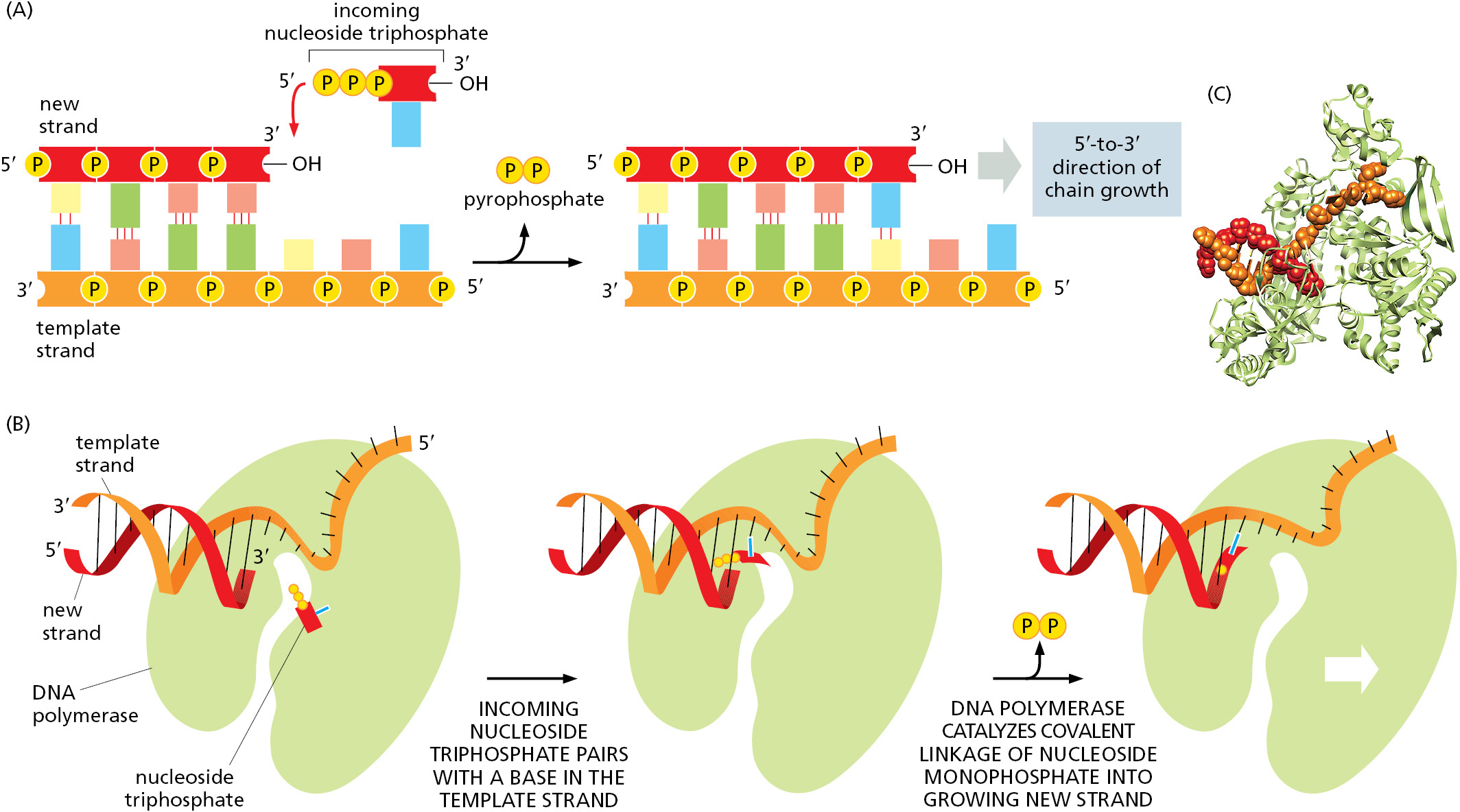
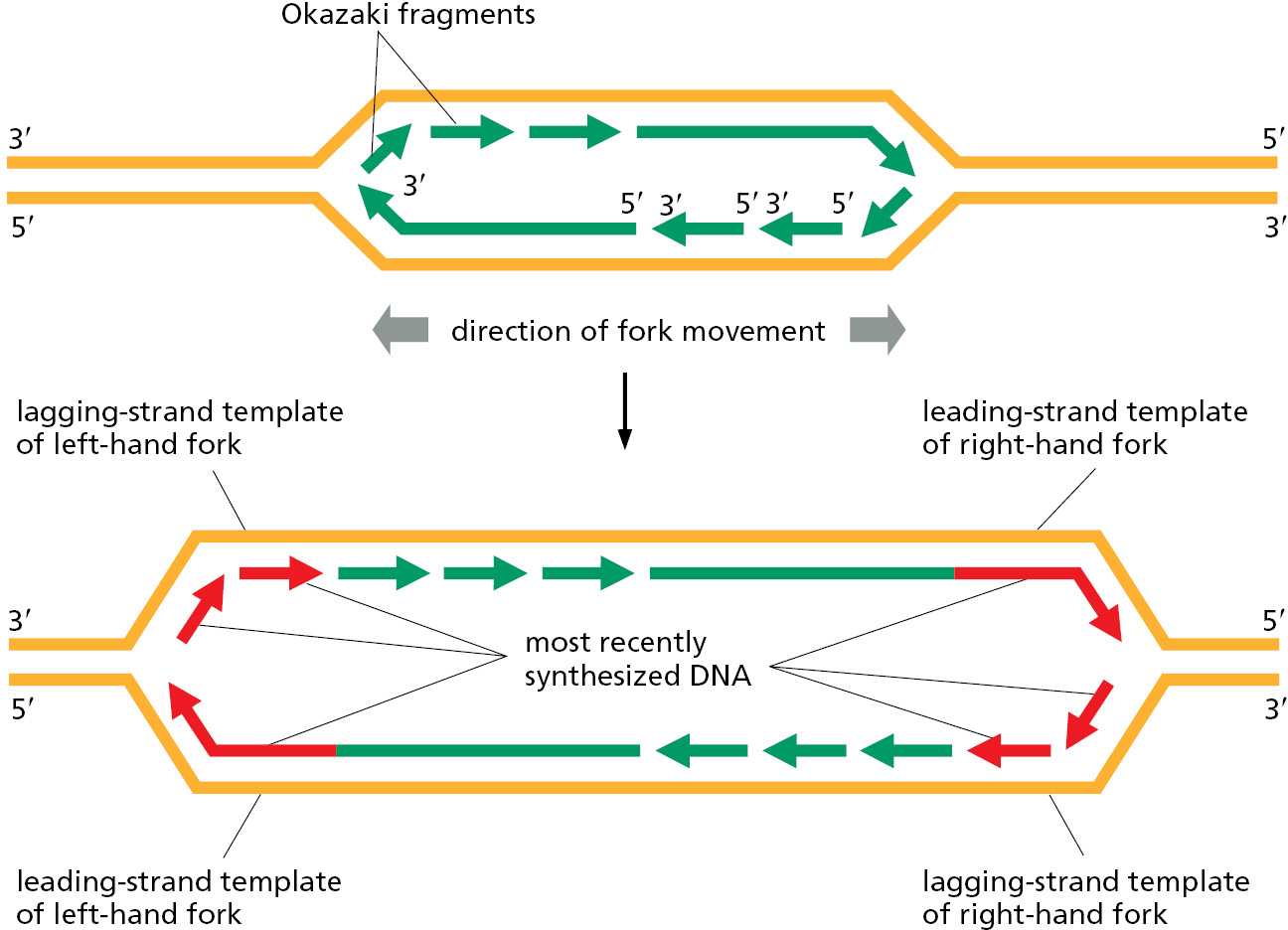
Initially, the simplest mechanism of DNA replication seemed to be the continuous growth of both new strands, nucleotide by nucleotide, at the replication fork as it moves from one end of a DNA molecule to the other. But because of the antiparallel orientation of the two DNA strands in the DNA double helix (see Figure 5–2), this mechanism would require one daughter strand to polymerize in the 5′-to-3′ direction and the other in the 3′-to-5′ direction. Such a replication fork would require two distinct types of DNA polymerase enzymes. However, as attractive as this model might seem, the DNA polymerases at replication forks can synthesize only in the 5′-to-3′ direction.
How, then, can a DNA strand grow in the 3′-to-5′ direction? The answer came from an experiment performed in the late 1960s. Researchers added highly radioactive 3H-thymidine to dividing bacteria for a few seconds, so that only the most recently replicated DNA—that just behind the replication fork—became radiolabeled. This experiment revealed the transient existence of pieces of DNA that were 1000–2000 nucleotides long, now commonly known as Okazaki fragments (named for their discoverer), at the growing replication fork. Similar replication intermediates were later found in eukaryotes, where they are only 100–200 nucleotides long. The Okazaki fragments were shown to be synthesized only in the 5′-to-3′ chain direction and to be joined together after their synthesis to create long DNA chains.
Each replication fork therefore has an asymmetric structure (Figure 5–7). The DNA daughter strand that is synthesized continuously is known as the leading strand. Its synthesis slightly precedes the synthesis of the daughter strand that is synthesized discontinuously, known as the lagging strand. For the lagging strand, the direction of nucleotide polymerization is opposite to the overall direction of DNA chain growth. The synthesis of this strand by a discontinuous “backstitching” mechanism means that DNA replication requires only the 5′-to-3′ type of DNA polymerase.
The High Fidelity of DNA Replication Requires Several Proofreading Mechanisms
As discussed at the beginning of the chapter, the fidelity of copying DNA during replication is such that only about one mistake occurs for every 1010 nucleotides copied. This accuracy is much higher than one would expect solely from the properties of complementary base-pairing. The standard complementary base pairs (see Figure 4–5) are not the only ones possible. For example, with small changes in helix geometry, two hydrogen bonds can form between G and T in DNA. In addition, rare configurations of the four DNA bases (known as tautomers) occur transiently in ratios of 1 part to 104 or 105. These forms mispair without a change in helix geometry: the rare tautomeric form of C pairs with A instead of G, for example.
If the DNA polymerase did nothing special when a mispairing occurred between an incoming deoxyribonucleoside triphosphate and the DNA template, the wrong nucleotide would often be incorporated into the new DNA chain, producing frequent mutations. The high fidelity of DNA replication, however, depends not only on the initial base-pairing, but also on several “proofreading” mechanisms that act sequentially to correct any initial mispairings that might have occurred.
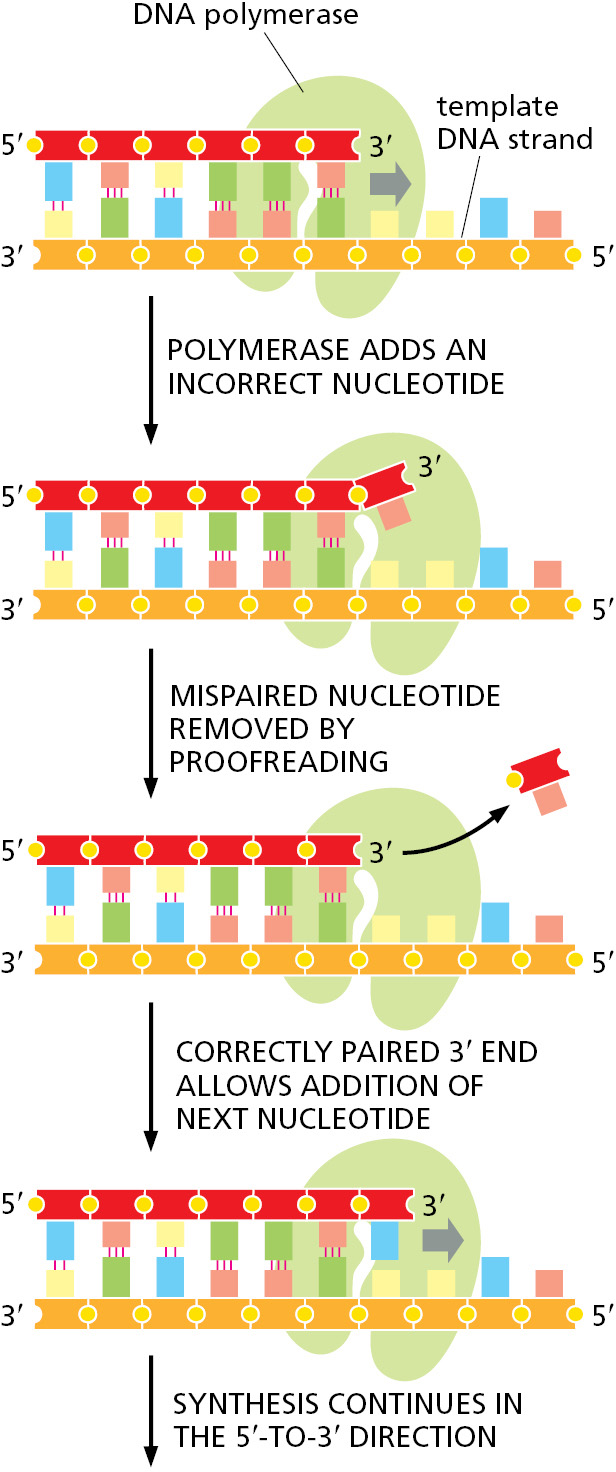
DNA polymerase performs the first proofreading step just before a new nucleotide is covalently added to the growing chain. After complementary nucleotide binding, but before the nucleotide is covalently added to the growing chain, the enzyme must undergo a conformational change in which its “grip” tightens around the active site. Because this change occurs more readily with correct than incorrect base-pairing, it allows the polymerase to “double-check” the exact base-pair geometry before it catalyzes the addition of the nucleotide. Incorrectly paired nucleotides are harder to add and therefore more likely to diffuse away before the polymerase can mistakenly add them.
The next error-correcting reaction, known as exonucleolytic proofreading, takes place immediately after those rare instances in which an incorrect nucleotide is covalently added to the growing chain. DNA polymerase enzymes are highly discriminating in the types of DNA chains they will elongate: they require a previously formed, base-paired 3′-OH end of a primer strand (see Figure 5–4). Those DNA molecules with a mismatched (improperly base-paired) nucleotide at the 3′-OH end of the primer strand are not effective as templates because the polymerase has difficulty extending such a strand. DNA polymerase molecules correct such a mismatched primer strand by means of a separate catalytic site (either in a separate subunit or in a separate protein domain of the polymerase molecule, depending on the polymerase). This 3′-to-5′ proofreading exonuclease clips off any unpaired or mispaired residues at the primer terminus, continuing until enough nucleotides have been removed to regenerate a correctly base-paired 3′-OH terminus that can prime DNA synthesis. In this way, DNA polymerase functions as a “self-correcting” enzyme that removes its own polymerization errors as it moves along the DNA (Figure 5–8 and Figure 5–9).
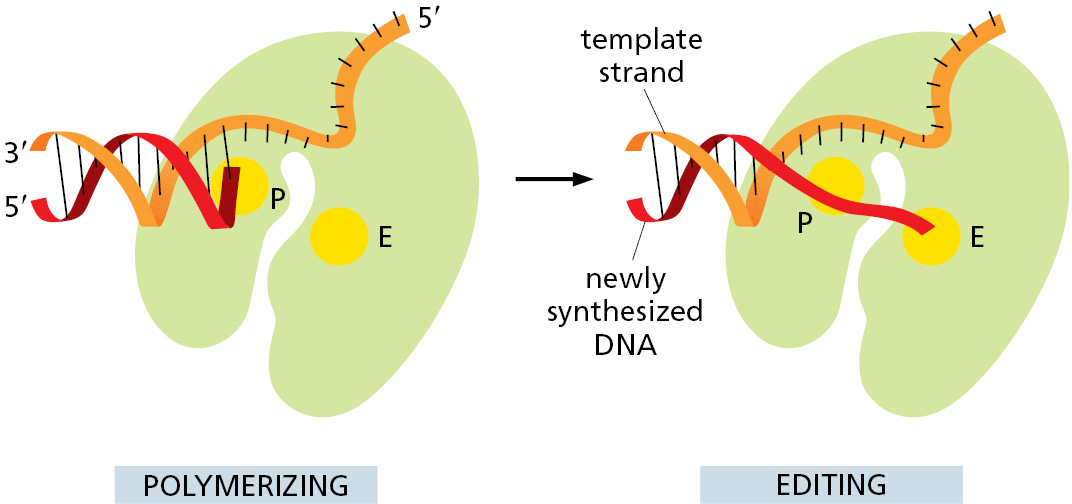
Because the self-correcting properties of DNA polymerase depend on its requirement for a perfectly base-paired primer terminus, it is apparently not possible for such an enzyme to start DNA synthesis de novo without an existing primer. By contrast, the RNA polymerase enzymes involved in gene transcription do not need such an efficient exonucleolytic proofreading mechanism: errors in making RNA are not passed on to the next generation, and the occasional defective RNA molecule that is produced has no long-term significance. As a result, RNA polymerases do not require a base-paired end 3′-OH for nucleotide addition and are able to start new polynucleotide chains without a primer.
On average, about one mistake is made for every 104 polymerization events both in RNA synthesis and in the separate process of translating mRNA sequences into protein sequences. This error rate is over 100,000 times greater than that in DNA replication, where, as we have seen, a series of proofreading processes makes the process unusually accurate (Table 5–1).
|
TABLE 5–1 The Three Steps That Give Rise to High-Fidelity DNA Synthesis |
|
|
Replication step |
Errors per nucleotide added |
|
5′→ 3′ polymerization |
1 in 105 |
|
3′→5′ exonucleolytic proofreading |
1 in 102 |
|
Strand-directed mismatch repair |
1 in 103 |
|
Combined |
1 in 1010 |
|
The third step, strand-directed mismatch repair, is described later in this chapter. For the polymerization step, “errors per nucleotide added” describes the probability that an incorrect nucleotide will be added to the growing chain. For the other two steps, “errors per nucleotide added” describes the probability that an error will not be corrected. Each step therefore reduces the chance of a final error by the factor shown. |
|
DNA Replication in the 5′-to-3′ Direction Allows Efficient Error Correction
The need for accuracy probably explains why DNA replication occurs only in the 5′-to-3′ direction. If there were a DNA polymerase that added deoxyribonucleoside triphosphates in the 3′-to-5′ direction, the growing 5′ end of the chain, rather than the incoming mononucleotide, would have to provide the activating triphosphate needed for the covalent linkage (see Figure 5–3). In this case, the mistakes in polymerization could not be simply hydrolyzed away, because the bare 5′ end of the chain thus created would immediately terminate DNA synthesis. It is therefore possible to correct a mismatched base only if it has been added to the 3′ end of a DNA chain. Although the backstitching mechanism for DNA replication seems complex, it preserves the 5′-to-3′ direction of polymerization that is required for exonucleolytic proofreading.
Despite these safeguards against DNA replication errors, DNA polymerases occasionally leave mistakes behind in the DNA that they produce. However, as we shall see later in this chapter, cells have yet another chance to correct these errors by a process called strand-directed mismatch repair. Before discussing this mechanism, however, we describe the other types of proteins that function at the replication fork as part of a large protein machine that replicates DNA.
A Special Nucleotide-polymerizing Enzyme Synthesizes Short RNA Primer Molecules
For the leading strand, a primer is needed only at the start of replication: once a replication fork is established, the DNA polymerase is continuously presented with a base-paired chain end on which to add new nucleotides. On the lagging side of the fork, however, each time the DNA polymerase completes a short DNA Okazaki fragment (which takes a few seconds), it must start synthesizing a completely new fragment at a site further along the template strand (see Figure 5–7). Each time this occurs, a special mechanism is required to produce a base-paired primer strand for the DNA polymerase to elongate. This requires an enzyme called DNA primase that uses ribonucleoside triphosphates to synthesize short RNA primers on the lagging strand (Figure 5–10). In eukaryotes, these primers are about 10 nucleotides long and are made at intervals of 100–200 nucleotides on the lagging strand. The synthesis of the leading strand also requires an RNA primer, but only at its very beginning.
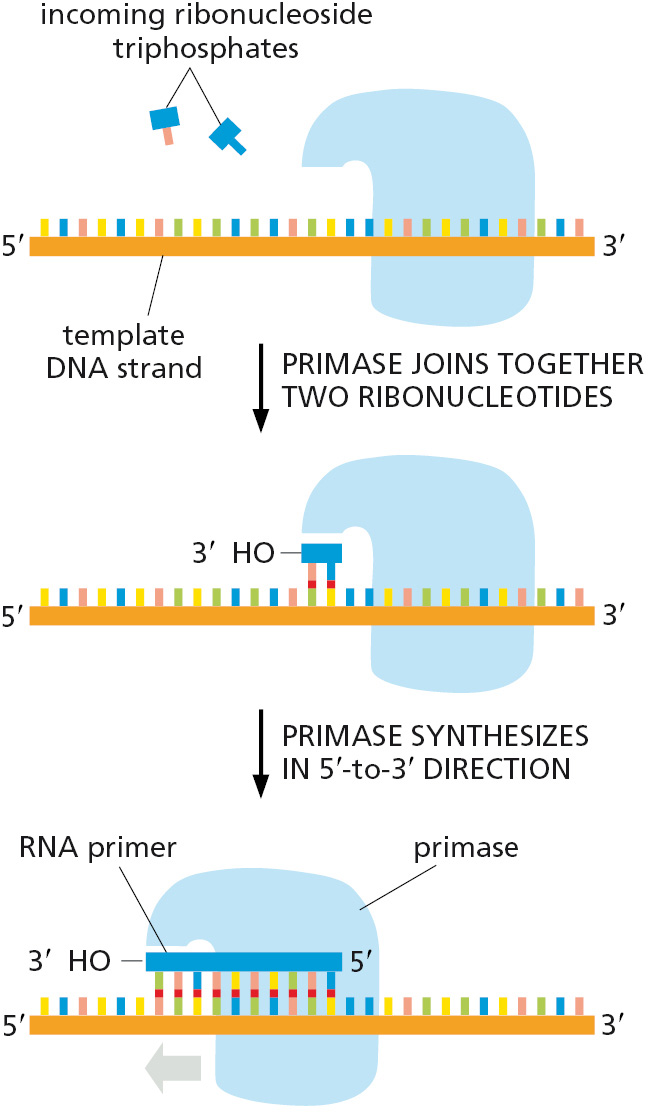
RNA was introduced in Chapter 1 and is described in detail in Chapter 6. Here, we note only that RNA is very similar in structure to DNA. A strand of RNA can form base pairs with a strand of DNA, generating a DNA–RNA hybrid double helix if the two nucleotide sequences are complementary. Thus, the same templating principle used for DNA synthesis guides the synthesis of RNA primers. Because an RNA primer contains a properly base-paired nucleotide with a 3′-OH group at one end, it can be elongated by the DNA polymerase at this end to begin an Okazaki fragment.
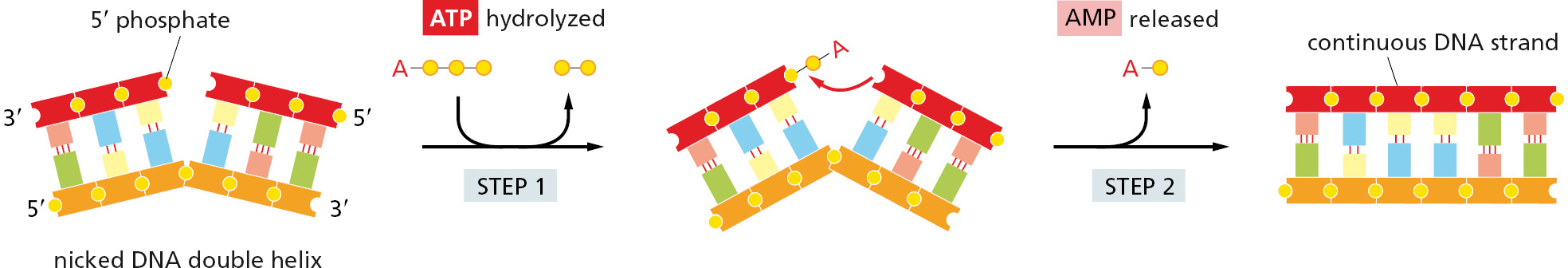
The synthesis of each Okazaki fragment ends when this DNA polymerase runs into the RNA primer attached to the 5′ end of the previous fragment. To produce a continuous DNA chain from the many DNA fragments made on the lagging strand, a special DNA repair system acts quickly to remove the RNA primers and replace them with DNA. An enzyme called DNA ligase then joins the 3′ end of the new DNA fragment to the 5′ end of the previous one to complete the process (Figure 5–11 and Figure 5–12).
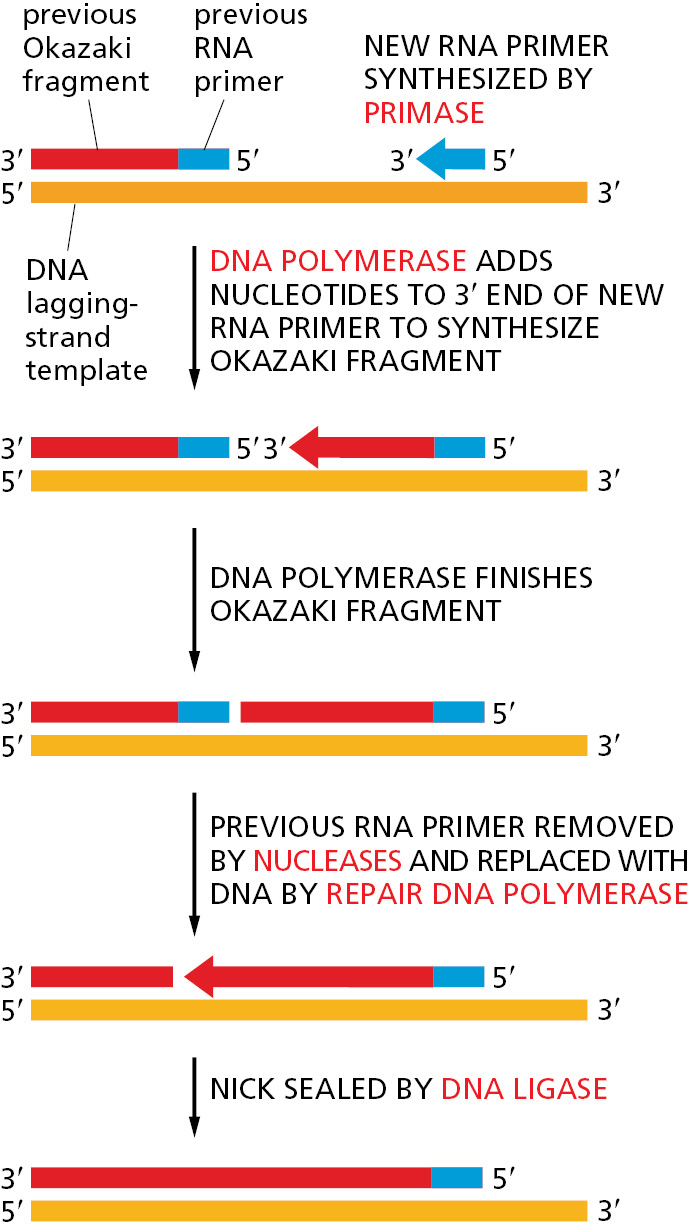
Why might an erasable RNA primer be used instead of a DNA primer? The argument that a self-correcting polymerase cannot start chains de novo also implies the converse: an enzyme that starts chains anew cannot be efficient at self-correction. Thus, any enzyme that primes the synthesis of Okazaki fragments will of necessity make a relatively inaccurate copy. If these inaccurate copies were allowed to remain, the resulting increase in the overall mutation rate would be enormous. It therefore seems likely that the use of RNA rather than DNA for priming brings a powerful advantage to the cell: the ribonucleotides in the primer automatically mark these sequences as “suspect copy” to be efficiently removed and replaced by DNA produced by a highly accurate DNA polymerase.
Special Proteins Help to Open Up the DNA Double Helix in Front of the Replication Fork
For DNA synthesis to proceed, the DNA double helix must be opened up ahead of the replication fork so that the incoming deoxyribonucleoside triphosphates can form base pairs with the template strand. The DNA double helix is very stable under physiological conditions: the base pairs are locked in place so strongly that it requires temperatures approaching that of boiling water to separate the two strands in a test tube. For this reason, two additional types of replication proteins—DNA helicases and single-strand DNA-binding proteins—are needed to open the double helix and present an appropriate single-stranded DNA template for the DNA polymerase to copy.
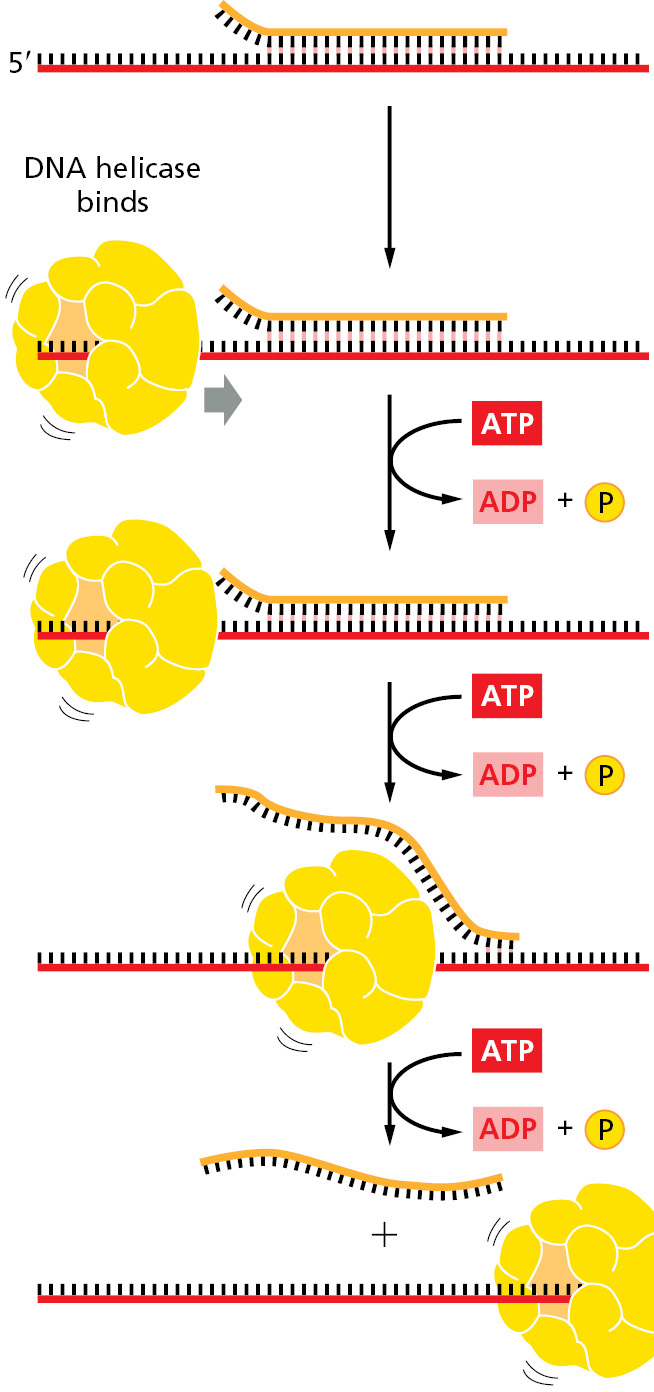
DNA helicases were first isolated as proteins that hydrolyze ATP when they are bound to single strands of DNA. As described in Chapter 3, the binding and hydrolysis of ATP can change the shape of a protein molecule in a cyclical manner that allows the protein to perform mechanical work. DNA helicases use this principle to propel themselves rapidly along a single DNA strand. When they encounter a region of double helix, they continue to move along their strand, thereby prying apart the helix ahead of them. This unidirectional movement can occur at rates of up to 1000 nucleotides per second (Figure 5–13 and Figure 5–14).
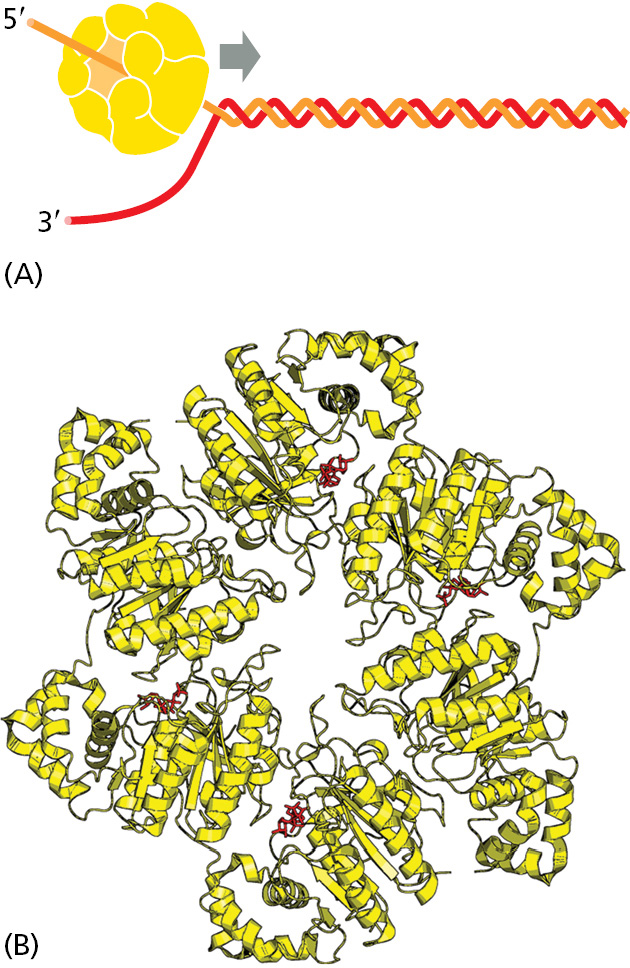
{kind=link}
The two strands of DNA have opposite polarities, and, in principle, a helicase could unwind the DNA double helix in front of a replication fork by moving either in the 5′-to-3′ direction along one strand or in the 3′-to-5′ direction along the other strand. In fact, both types of DNA helicase exist. In the best-understood replication systems in bacteria, a helicase moving 5′-to-3′ along the lagging-strand template has the predominant role.
Single-strand DNA-binding (SSB) proteins bind tightly and cooperatively to the single-stranded DNA that is produced by helicases. Through cooperative binding, SSB proteins coat and straighten out all regions of single-stranded DNA, thereby preventing the formation of the short hairpin helices that otherwise form in these single strands (Figure 5–15 and Figure 5–16). These regions occur routinely on the lagging-strand template, and if not removed, they can impede the DNA synthesis catalyzed by DNA polymerase.
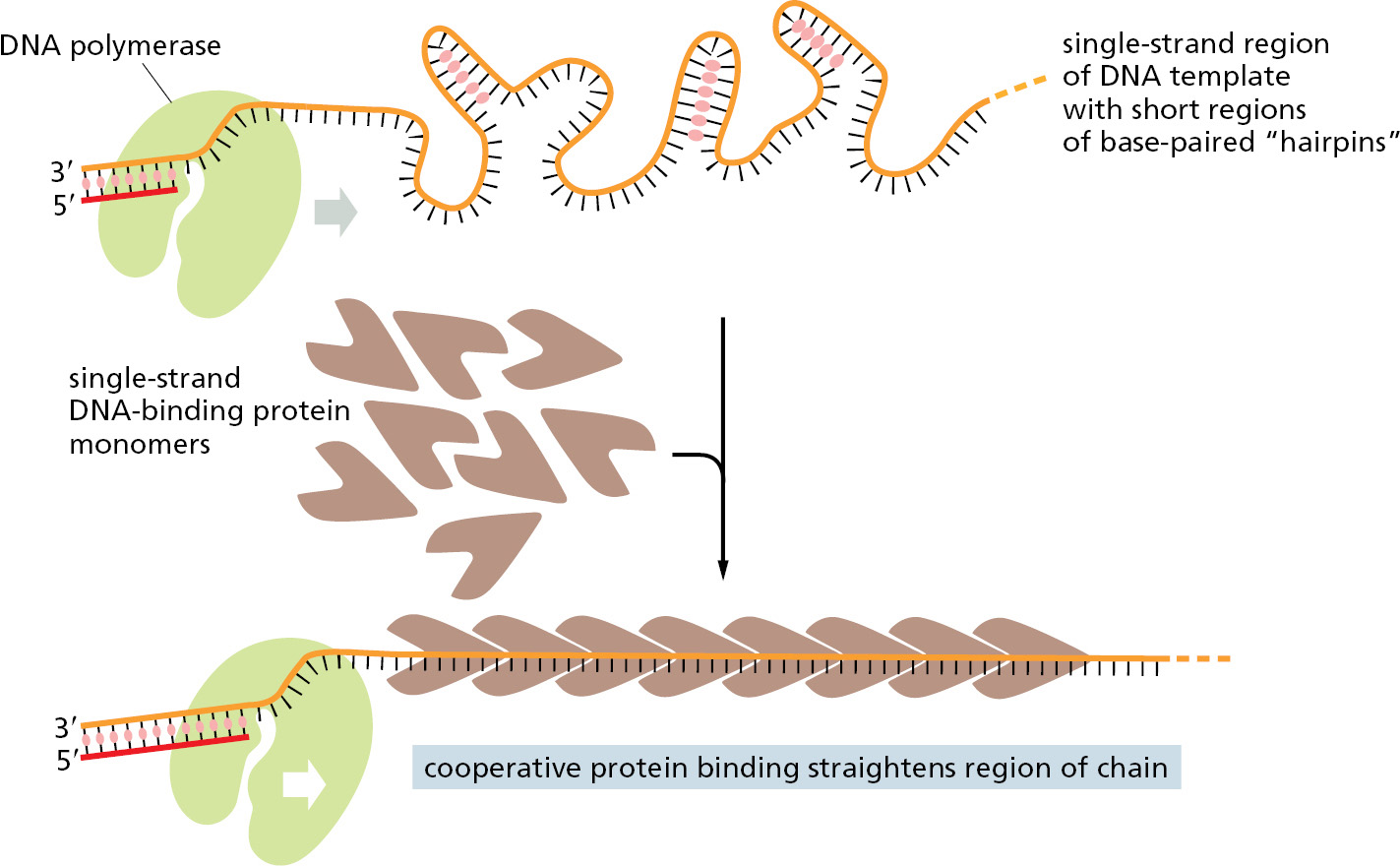
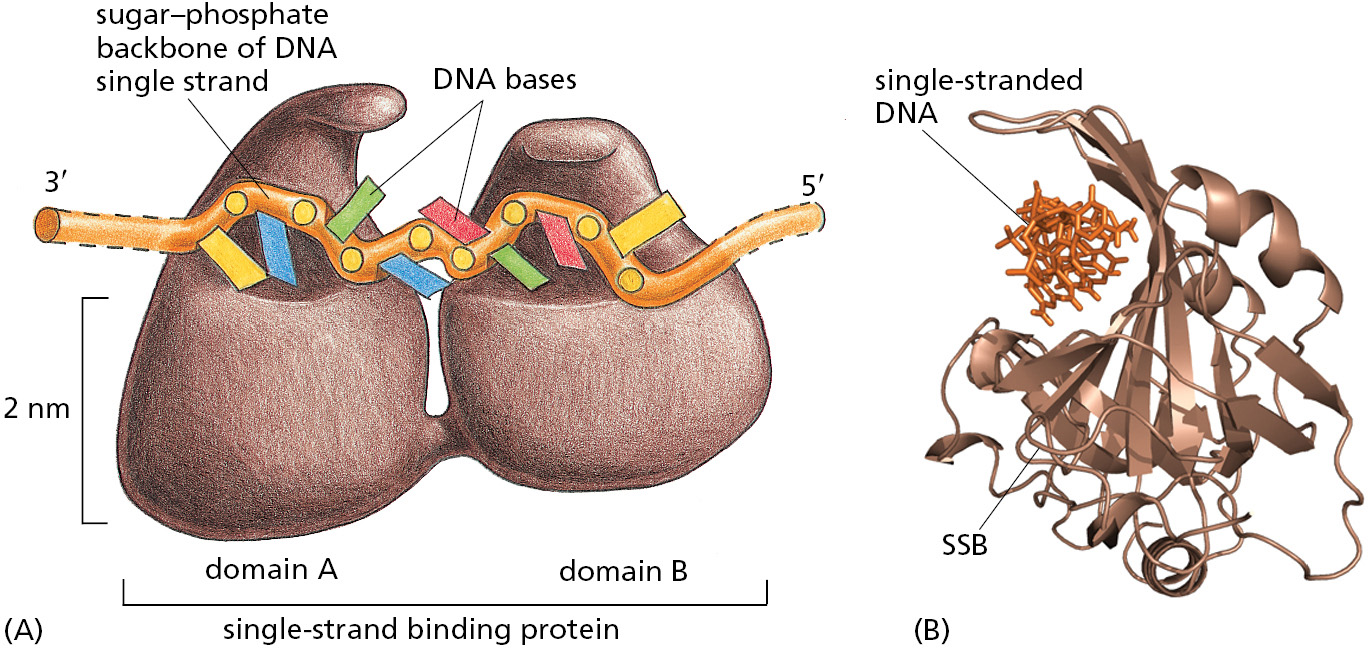
A Sliding Ring Holds a Moving DNA Polymerase onto the DNA
On their own, most DNA polymerase molecules will synthesize only a short string of nucleotides before falling off the DNA template. However, an accessory protein (called PCNA in eukaryotes) forms a sliding clamp that keeps the polymerase firmly on the DNA when it is moving but releases the polymerase as soon as it runs into a double-strand region of DNA.
How can a sliding clamp prevent the polymerase from dissociating without impeding the polymerase’s rapid movement along DNA? The three-dimensional structure of the clamp protein revealed that it forms a large ring around the DNA double helix. One face of the ring binds to the back of the DNA polymerase, and the whole ring slides freely along the DNA as the polymerase moves. The assembly of the clamp around the DNA requires a special protein complex, the clamp loader, that can open and close the ring in a regulated manner.
The moving DNA polymerase is tightly bound to the clamp, and, on the leading strand, the two remain associated for a very long time. The DNA polymerase on the lagging-strand template also makes use of the clamp, but each time the polymerase reaches the 5′ end of the preceding Okazaki fragment, the polymerase releases itself from the clamp and dissociates from the template. With the help of the clamp loader, which hydrolyzes ATP as it loads a new clamp onto a primer–template junction (Figure 5–17), this lagging-strand polymerase molecule then associates with the new clamp that is assembled on the RNA primer of the next Okazaki fragment.
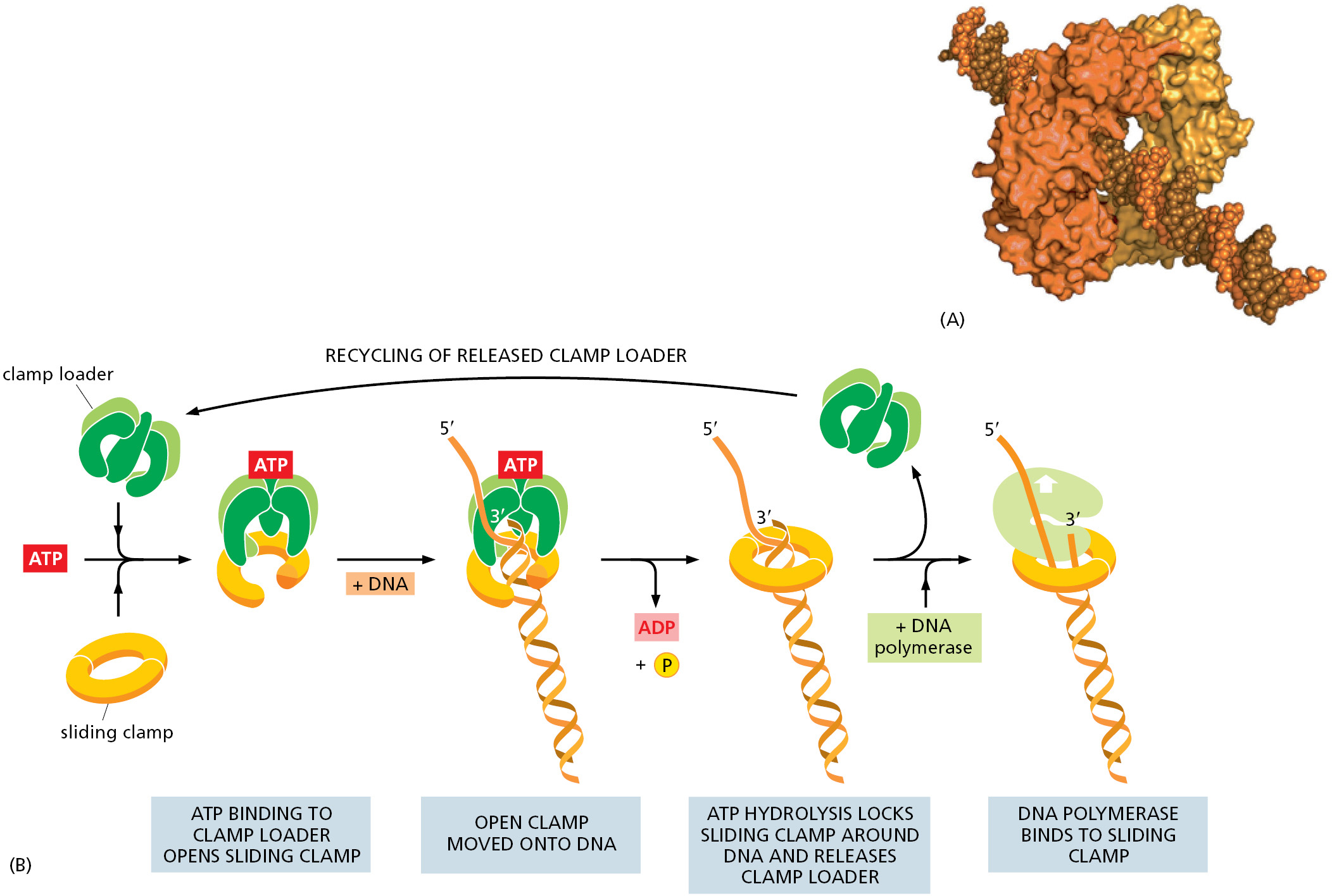
{kind=link}
The Proteins at a Replication Fork Cooperate to Form a Replication Machine
Although we have discussed DNA replication as though it were performed by a set of proteins all acting independently, in reality most of these proteins are held together in a large and orderly multienzyme complex that rapidly synthesizes DNA. This complex can be likened to a tiny sewing machine composed of protein parts and powered by nucleoside triphosphate hydrolysis. Like a sewing machine, the replication complex probably remains stationary with respect to its immediate surroundings; the DNA can be thought of as a long strip of cloth being rapidly threaded through it. Although the replication complex has been most intensively studied in E. coli and several of its viruses, a very similar complex also operates in eukaryotes, as we shall see below.
How the different proteins at the replication fork work together in bacteria is shown in Figure 5–18. At the front of the replication fork, DNA helicase opens the DNA helix. Several identical DNA polymerase molecules work at the fork, one on the leading strand and two on the lagging strand. Whereas the DNA polymerase molecule on the leading strand can operate in a continuous fashion, the DNA polymerase molecules on the lagging-strand alternate at short intervals, using the short RNA primers made by DNA primase to begin each Okazaki fragment. The close association of all these protein components increases the efficiency of replication, and it is made possible by a folding back of the lagging strand as shown in the figure. This arrangement facilitates the loading of the polymerase clamp each time that an Okazaki fragment is synthesized: the clamp loader and the lagging-strand DNA polymerase molecule are kept in place at the replication fork even when they detach from their DNA template. The replication proteins are thus linked together into a single large unit (total molecular mass >106 daltons), enabling DNA to be synthesized on both sides of the replication fork in a coordinated and efficient manner.
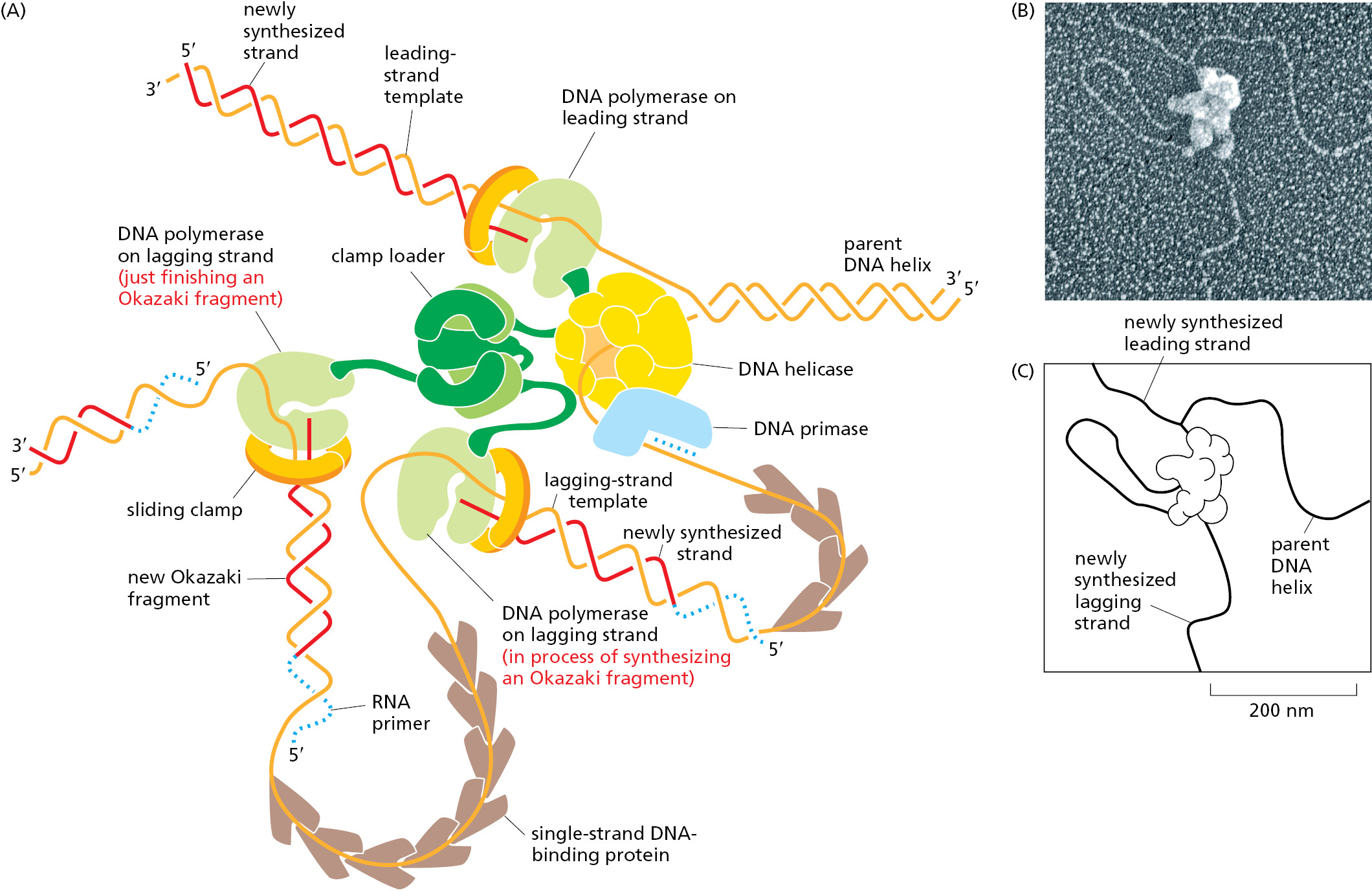
{kind=link}
On the lagging strand, the DNA replication machine leaves behind a series of unsealed Okazaki fragments, which still contain the RNA that primed their synthesis at their 5′ ends. As discussed earlier, this RNA is removed, and the resulting gap is filled in by DNA repair enzymes that operate behind the replication fork (see Figure 5–11).
DNA Replication Is Fundamentally Similar in Eukaryotes and Bacteria
Much of what we know about DNA replication was first derived from studies of purified bacterial and bacteriophage multienzyme systems capable of DNA replication in vitro. The development of these systems in the 1970s was greatly facilitated by the prior isolation of mutants in a variety of replication genes; these mutants were exploited to identify and purify the corresponding replication proteins. The first eukaryotic replication system that accurately replicated DNA in vitro was described in the mid-1980s, and mutations in genes encoding nearly all of the replication components have now been isolated and analyzed in the yeast Saccharomyces cerevisiae. As a result, much is known about the detailed enzymology of DNA replication in eukaryotes, and it is clear that the fundamental features of DNA replication—including replication-fork geometry and the use of 5′→3′ DNA polymerases, helicases, clamps, clamp loaders, and single-strand binding proteins—are similar.
However, there are some important differences in how bacteria and eukaryotes replicate their DNA. Perhaps most important, eukaryotes use three different kinds of DNA polymerase at each replication fork (Figure 5–19). Polymerase ε (Polε) synthesizes the leading strand, whereas Polα and Polδ synthesize the lagging-strand Okazaki fragments. Each type of polymerase has special properties that make it well suited for its job. Polε binds to both the sliding clamp and the replicative helicase, allowing it to synthesize very long stretches of leading-strand DNA without dissociating. Polα includes DNA primase as one of its subunits, which begins all new chains by synthesizing a short length of RNA. This RNA is extended by a different subunit of Polα, which adds only about 20 nucleotides of DNA before dissociating. Finally, Polδ, which is loaded in conjunction with a sliding clamp, takes over and completes synthesis of each Okazaki fragment to produce a total length of about 200 nucleotides.
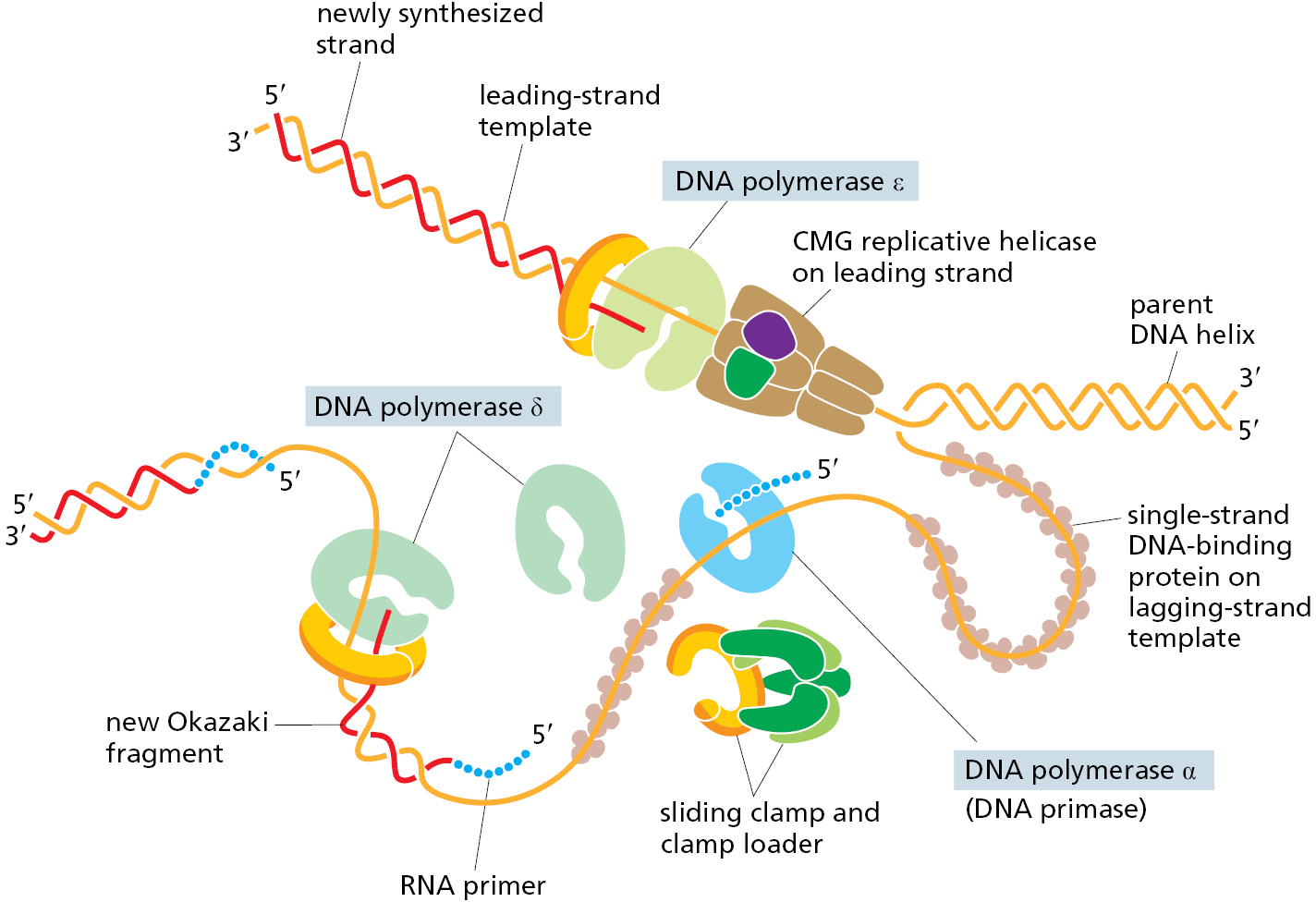
{kind=link}
The use of three different kinds of DNA polymerase at the replication fork is part of a trend toward higher complexity observed for eukaryotic DNA replication compared to that of bacteria. As another example, the eukaryotic single-strand binding protein is formed from three different subunits, while only a single subunit is found in bacteria. Likewise, the eukaryotic replicative helicase (known as the CMG helicase) is composed of 11 different protein subunits, while the bacterial enzyme is a hexamer of 6 identical subunits. We do not know why the eukaryotic replication machinery is so much more complex than that of bacteria; however, there are several possibilities. In eukaryotes, DNA replication must be coordinated with the elaborate process of mitosis; it must also deal with DNA packaged into nucleosomes, topics we discuss in the next part of the chapter. It is also possible that the difference in complexity between bacteria and eukaryotes largely reflects evolutionary pressure for bacteria to make do with fewer genes.
Another important distinction between eukaryotic and bacterial replication protein complexes lies in the detailed structures of their individual protein components. With the exception of the sliding clamp, the replication proteins in bacteria have completely different structures and amino acid sequences than those of their eukaryotic counterparts. The simplest interpretation of this surprising fact is that, over hundreds of millions of years, the DNA replication machinery in eukaryotes and bacteria evolved independently, yet converged on the same basic mechanisms. This situation is in contrast to other fundamental processes in the cell, such as transcription and translation, where the fundamental components (RNA polymerase and the ribosome) are very similar between bacteria and eukaryotes—and where the structures are conserved from an ancient, common ancestor.
A Strand-directed Mismatch Repair System Removes Replication Errors That Remain in the Wake of the Replication Machine
Because bacteria such as E. coli are capable of dividing once every 30 minutes, it is relatively easy to screen large populations to find a rare mutant cell that is altered in a specific process. One interesting class of mutants consists of those with alterations in so-called mutator genes, which greatly increase the rate of spontaneous mutation. Not surprisingly, one such mutant makes a defective form of the 3′-to-5′ proofreading exonuclease that is a part of the DNA polymerase enzyme (see Figures 5–8 and 5–9). The mutant DNA polymerase no longer proofreads effectively, and many replication errors that would otherwise have been removed accumulate in the DNA.
The study of other E. coli mutants exhibiting abnormally high mutation rates uncovered an additional proofreading system, common to all cells on Earth, that removes those rare replication errors that were made by the polymerase and missed by its proofreading exonuclease. These errors leave mismatched base pairs behind the replication fork, which are subsequently recognized and corrected by a strand-directed mismatch repair system. This system picks out mismatches from normal DNA by monitoring their potential to distort the DNA double helix, which is greatly increased by the misfit between noncomplementary base pairs. However, if the repair system simply recognized a mismatch in newly replicated DNA and randomly corrected one of the two mismatched nucleotides, it would mistakenly “correct” the original template strand to match the error exactly half the time, thereby failing to lower the overall error rate. To be effective, such a proofreading system must be able to remove only the nucleotide on the newly synthesized strand, where the error occurred.
The strand-distinction mechanism used by the mismatch proofreading system in E. coli depends on the methylation of selected A residues in the DNA. Methyl groups are added to all A residues in the sequence GATC, but not until some time after the GATC has been synthesized. As a result, the only unmethylated GATC sequences lie in the newly synthesized strands just behind a replication fork. The recognition of these unmethylated GATCs (which are base-paired to methylated GATCs) allows the new DNA strands to be transiently distinguished from old ones, as required if their mismatches are to be selectively removed. The five-step error-correction process involves recognition of a mismatch, identification of the newly synthesized strand, excision of the portion containing the misincorporated nucleotide, resynthesis of the excised segment using the old strand as a template, and ligation to seal the DNA backbone. This strand-directed mismatch repair system reduces the number of errors made during DNA replication by an additional factor of 100–1000 (see Table 5–1, p. 260).
A similar mismatch proofreading system functions in eukaryotic cells, but it uses a different way to distinguish the newly synthesized DNA strands from the parent strands. On the lagging strand, the newly synthesized DNA will contain transient single-strand gaps before the series of Okazaki fragments are processed and ligated together. Each gap will usually carry a sliding clamp, which remains on the DNA after the DNA polymerase has dissociated from it to begin the next fragment. Together, the clamp and the single-strand break signal to the mismatch repair proteins to correct the mismatch using the parent DNA strand as the template (Figure 5–20).
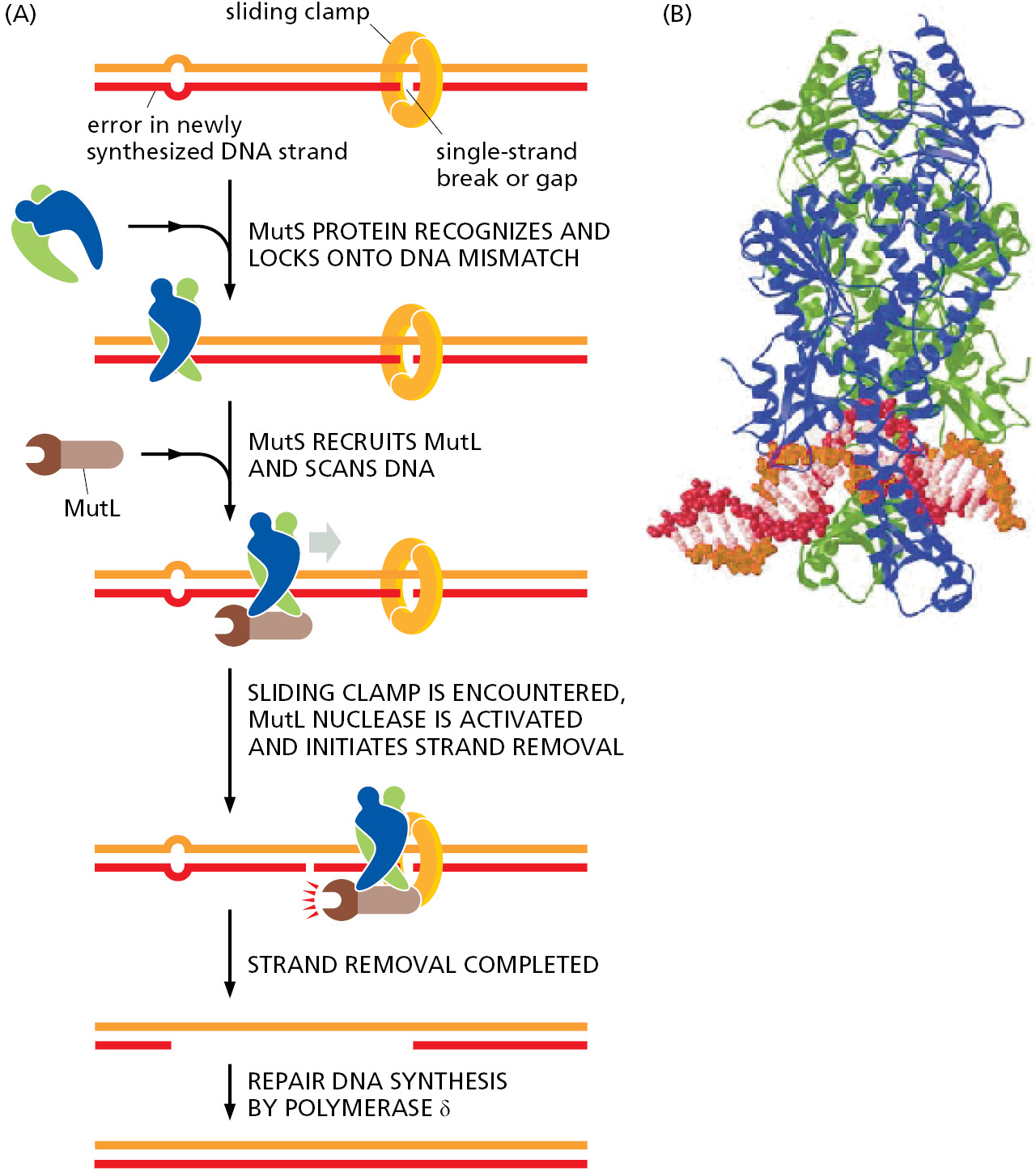
The two faces of the clamp differ, and the clamp loader always loads the clamp in the same orientation with respect to the 3′ end of the previously synthesized Okazaki fragment. Because all the clamps on the DNA “face” in the same direction relative to the replication process, the oriented clamps can be used by the mismatch repair machinery to distinguish newly synthesized DNA from parent DNA. It is not known for certain how strand discrimination occurs on the leading strand (where gaps in newly synthesized DNA should be rare), but because oriented sliding clamps are also left behind by the leading-strand polymerase, they can signal old from new DNA in the same way that they do on the lagging strand. The recent discovery of a correction system that removes misincorporated ribonucleotides suggests a further possibility for distinguishing newly synthesized DNA from parent DNA, as we discuss in the next section.
Mismatch correction is crucial for all cells; its importance for humans is seen in individuals who inherit one defective copy of a mismatch repair gene (along with a functional gene on the other copy of the chromosome). These individuals have a marked predisposition for certain types of cancers. For example, in a type of colon cancer called hereditary nonpolyposis colorectal cancer (HNPCC), a spontaneous deleterious mutation of the one functional gene will produce a clone of somatic cells that, because they are deficient in mismatch proofreading, accumulate mutations unusually rapidly. Because most cancers arise in cells that have accumulated many mutations (as discussed in Chapter 20), cells deficient in mismatch proofreading have a greatly enhanced chance of becoming cancerous. Fortunately, most of us inherit two good copies of each gene that encodes a mismatch proofreading protein; this protects us, because it is highly unlikely that both copies will become mutated in the same cell.
The Accidental Incorporation of Ribonucleotides During DNA Replication Is Corrected
We have seen that cells have several ways to correct mistakes where the wrong deoxynucleotide has been incorporated in newly replicated DNA. Occasionally, however, DNA polymerases make a different kind of mistake, one that is not caused by improper base-pairing: in this case, they accidently incorporate a ribonucleotide instead of a deoxyribonucleotide. These molecules differ by a single –OH group in the sugar portion of the nucleotide. Yet, when incorporated into DNA, they weaken the DNA chain at that point, rendering it highly susceptible to breakage. If left unrepaired, these “weak links” would cause high mutation rates and genome rearrangements. Even if it does not cause a break, an incorporated ribonucleotide distorts the DNA double helix and can stall some polymerases during the next cycle of DNA replication.
Although DNA polymerases much prefer deoxyribonucleotides over ribonucleotides (by a factor of about a million), the concentration of ribonucleotides in the cell is much higher than that of their deoxy counterparts, as much as 500-fold for ATP, which has many different uses in the cell. This concentration imbalance means that a ribonucleotide is accidentally incorporated approximately once per several thousand nucleotides of DNA synthesized. These mistakes are corrected by specific nucleases that cleave the DNA chain when they encounter a ribonucleotide, leading to the excision of the ribonucleotide and its replacement by DNA, much in the same way that RNA primers are replaced by DNA to complete lagging-strand synthesis (see Figure 5–11). Because this repair process produces gaps only in newly synthesized DNA, it has been proposed that these transient lesions help the mismatch repair system “know” which strand to repair; in particular, these cues may be especially important on the leading strand.
DNA Topoisomerases Prevent DNA Tangling During Replication
As a replication fork moves along double-stranded DNA, it creates what has been called the “winding problem.” The two parent strands that are wound around each other must be unwound and separated for replication to occur. For every 10 nucleotide pairs replicated at the fork, one complete turn of the parent double helix must be unwound. In principle, this unwinding can be achieved by rapidly rotating the entire chromosome ahead of a moving fork; however, this is energetically highly unfavorable (particularly for long chromosomes). Instead, the DNA in front of a replication fork becomes overwound (Figure 5–21). This overwinding is continually relieved by enzymes known as DNA topoisomerases.
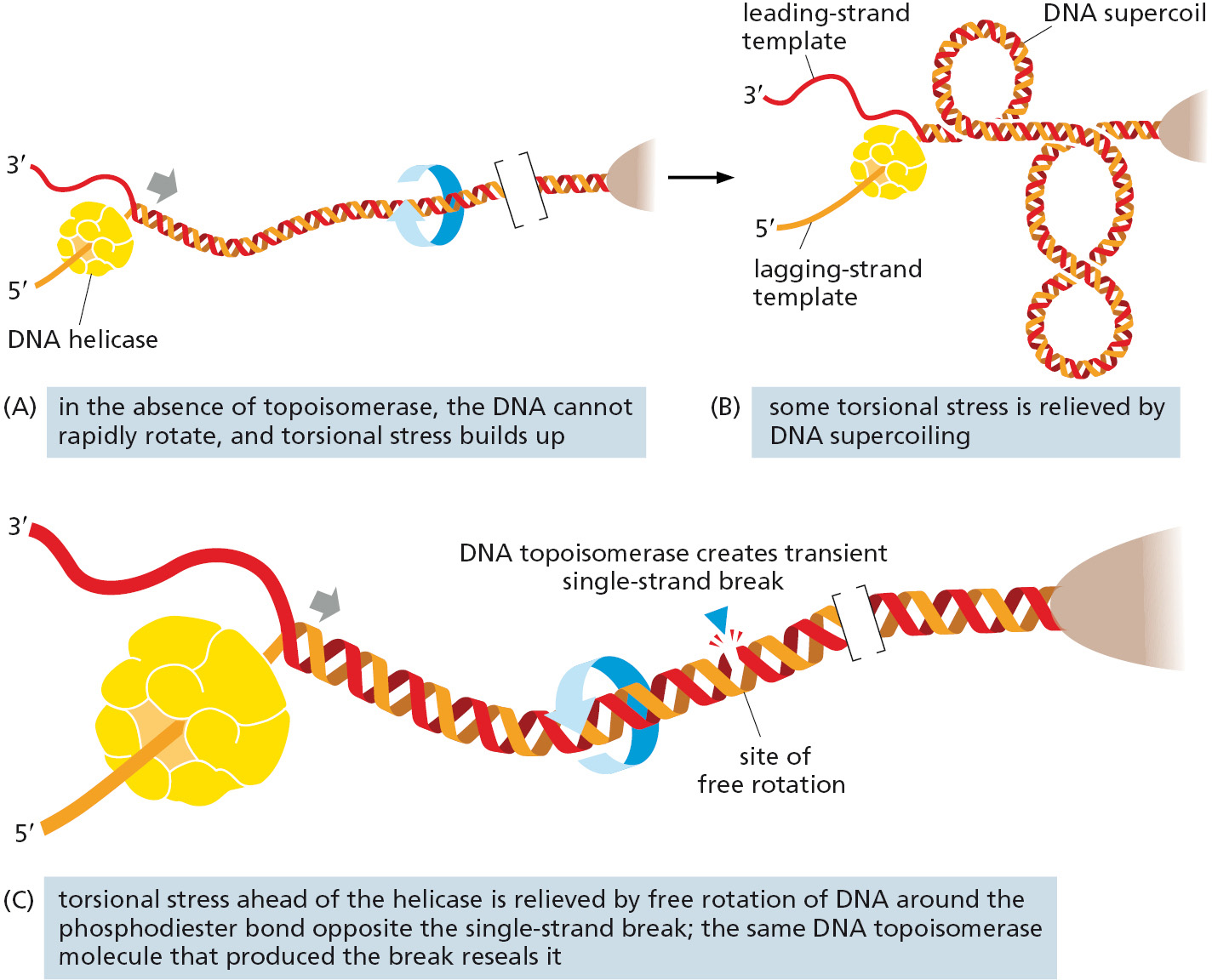
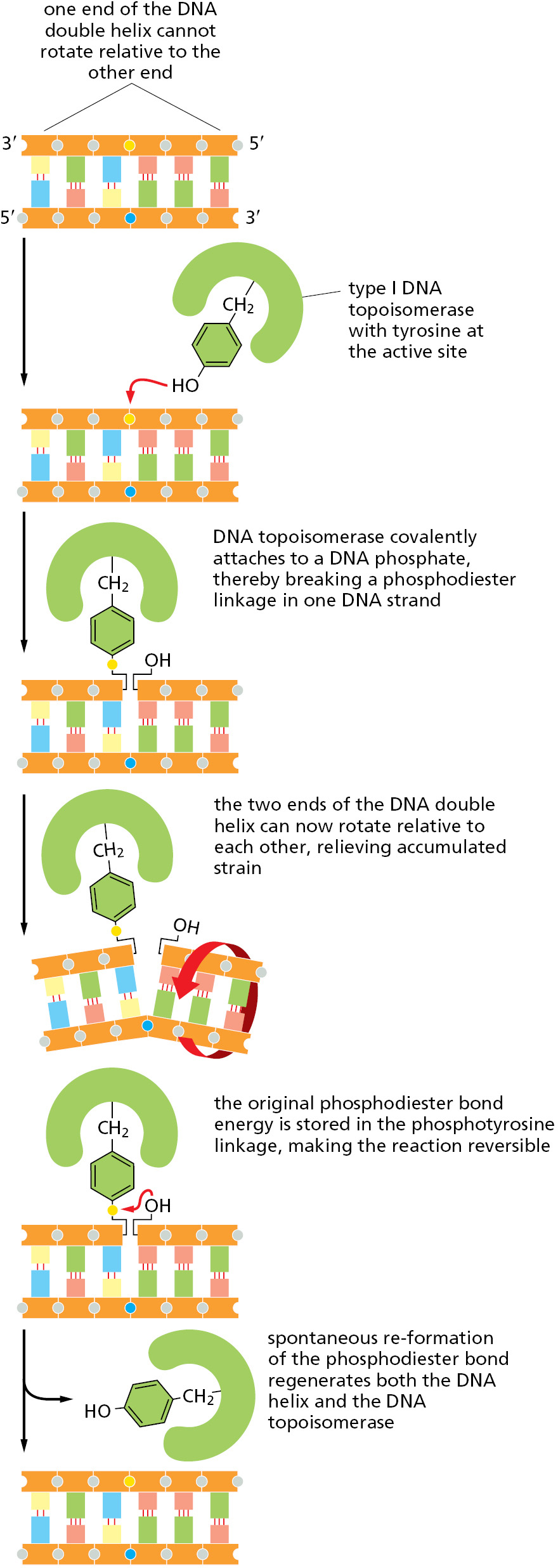
A DNA topoisomerase can be viewed as a reversible nuclease that adds itself covalently to a DNA backbone phosphate, thereby breaking a phosphodiester bond in a DNA strand. This reaction is reversible, and the phosphodiester bond re-forms as the protein leaves.
One type of topoisomerase, called topoisomerase I, produces a transient single-strand break; this break in the phosphodiester backbone allows the two sections of DNA helix on either side of the nick to rotate freely relative to each other, using the phosphodiester bond in the strand opposite the nick as a swivel point (Figure 5–22). Any tension in the DNA helix will drive this rotation in the direction that relieves the tension. As a result, DNA replication can occur with the rotation of only a short length of helix—the part just ahead of the fork. Because the covalent linkage that joins the DNA topoisomerase protein to a DNA phosphate retains the energy of the cleaved phosphodiester bond, resealing is rapid and does not require additional energy input. In this respect, the rejoining mechanism differs from that catalyzed by the enzyme DNA ligase, discussed previously (see Figure 5–12).
A second type of DNA topoisomerase, topoisomerase II, forms a covalent linkage to both strands of the DNA helix at the same time, making a transient double-strand break in the helix. These enzymes are activated by sites on chromosomes where two double helices cross over each other, such as those generated by supercoiling in front of a replication fork (see Figure 5–21B). As illustrated in Figure 5–23, once a topoisomerase II molecule binds to such a crossing site, the protein uses ATP hydrolysis to perform the following set of reactions: (1) it breaks one double helix reversibly to create a DNA “gate”; (2) it causes the second, nearby double helix to pass through this opening; and (3) it then reseals the break and dissociates from the DNA. At crossover points generated by supercoiling, passage of the double helix through the gate occurs in the direction that will reduce supercoiling. In this way, type II topoisomerases—like type I topoisomerases—can relieve the overwinding tension generated in front of a replication fork.
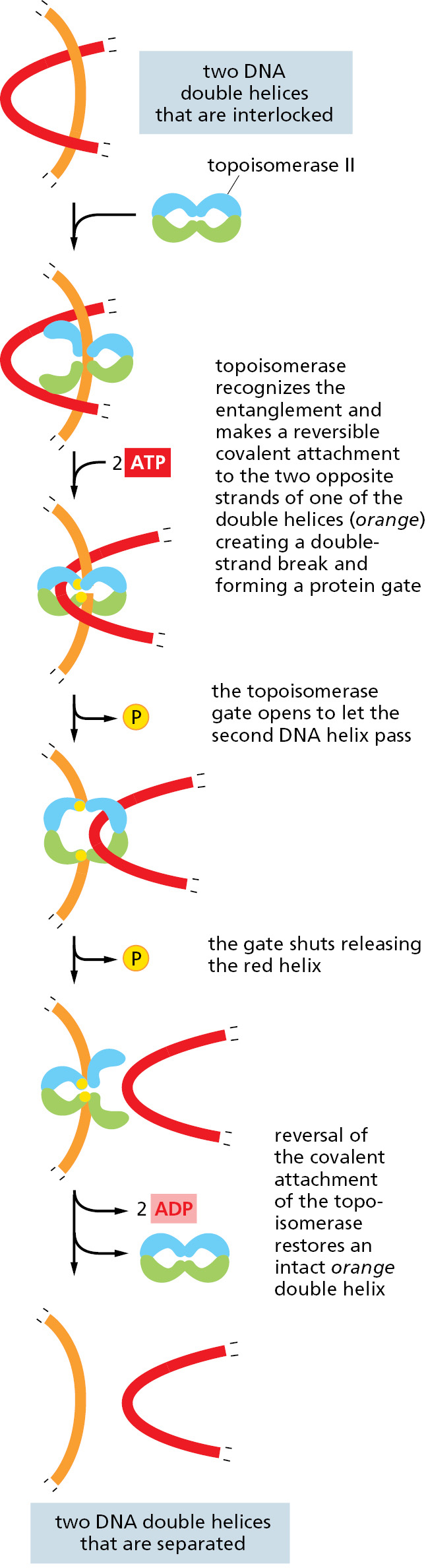
Their reaction mechanism also allows type II DNA topoisomerases to efficiently separate any intertwined DNA molecules. This ability of topoisomerase II is especially important for preventing the severe DNA tangling problems that would otherwise arise from DNA replication. This role is nicely illustrated by mutant yeast cells that produce, in place of the normal topoisomerase II, a version that is inactive above 37°C. When the mutant cells are warmed to this temperature, their daughter chromosomes remain intertwined after DNA replication and are unable to separate. The enormous usefulness of topoisomerase II for untangling chromosomes before mitosis begins can readily be appreciated by anyone who has struggled to remove a severe tangle from a fishing line—or from a large ball of thread—without the aid of scissors.
Summary
DNA replication takes place at a Y-shaped structure called a replication fork. Self-correcting DNA polymerase enzymes catalyze nucleotide polymerization in a 5′-to-3′ direction, copying a DNA template strand with remarkable fidelity. Because the two strands of a DNA double helix are antiparallel, this 5′-to-3′ DNA synthesis can take place continuously on only one of the strands at a replication fork (the leading strand). On the lagging strand, short DNA fragments must be made by a “backstitching” process. Because the self-correcting DNA polymerases cannot start a new chain, these lagging-strand DNA fragments are primed by short RNA primer molecules that are subsequently erased and replaced with DNA.
DNA replication requires the cooperation of many proteins. These include (1) DNA polymerases and DNA primases to catalyze nucleoside triphosphate polymerization; (2) DNA helicases and single-strand DNA-binding (SSB) proteins to help in opening up the DNA helix so that it can be copied; (3) clamps and clamp loaders to enable DNA polymerases to copy longer stretches of DNA; (4) DNA ligases and enzymes that degrade RNA primers to seal together the discontinuously synthesized lagging-strand DNA fragments; and (5) DNA topoisomerases to help to relieve helical winding and DNA tangling problems. Many of these proteins associate with each other at a replication fork to form a highly efficient “replication machine,” through which the activities and spatial movements of the individual components are coordinated.
The self-correcting DNA polymerases make mistakes only rarely when copying DNA; when they do, a variety of enzymes inspect the DNA shortly after it is made and correct any mishaps. Given the number of proteins dedicated to the task, copying DNA with extreme accuracy is clearly of great importance to all cells on Earth.
Glossary
- DNA polymerase
- Enzyme that synthesizes DNA by joining nucleotides together using a DNA template as a guide; its substrates are the four nucleoside triphosphates: A, G, C, and T.
- replication fork
- Y-shaped region of a replicating DNA molecule; the point at which the two strands of the parent DNA helix are being separated and the daughter strands are being formed.
- leading strand
- One of the two newly synthesized strands of DNA found at a replication fork. The leading strand is made by continuous synthesis in the 5′-to-3′ direction.
- lagging strand
- One of the two newly synthesized strands of DNA found at a replication fork. The lagging strand is made in discontinuous lengths that are later joined covalently.
- DNA primase
- Enzyme that synthesizes a short strand of RNA on a DNA template, producing an RNA primer for DNA synthesis.
- RNA primer
- Short stretch of RNA synthesized on a DNA template. It is required by DNA polymerases to start their DNA synthesis.
- DNA ligase
- Enzyme that joins the ends of two strands of DNA together with a covalent bond to produce one continuous DNA strand.
- DNA helicase
- Enzyme that harnesses ATP hydrolysis energy to open a region of the DNA helix into its single strands as an aid to DNA replication or DNA repair.
- single-strand DNA-binding (SSB) protein
- Protein that binds to the single strands of the opened-up DNA double helix, preventing helical structures from re-forming while the DNA is being replicated.
- sliding clamp
- Ring-shaped protein complex that holds the DNA polymerase on DNA during DNA replication.
- clamp loader
- Protein complex that utilizes ATP hydrolysis to load the sliding clamp onto a primer–template junction in the process of DNA replication.
- strand-directed mismatch repair
- A proofreading system that removes DNA replication errors missed by the DNA polymerase proofreading exonuclease. It detects the DNA helix distortion from noncomplementary base pairs and then excises the mismatch in the newly synthesized strand specifically; the excised DNA segment is then replaced using the old strand as a template.
- DNA topoisomerase
- Enzyme that binds to DNA and reversibly breaks a phosphodiester bond in one or both strands. Topoisomerase I creates transient single-strand breaks, allowing the double helix to swivel and relieving superhelical tension. Topoisomerase II creates transient double-strand breaks, allowing one double helix to pass through another and thus resolving knots and tangles.