For proteins, form and function are inextricably linked. Dictated by the surface topography of a protein’s side chains, this union of structure, chemistry, and activity gives proteins the extraordinary ability to orchestrate the large number of dynamic processes that occur in cells. But the fundamental question remains: How do proteins actually work? In this section, we will see that the activity of proteins depends on their ability to bind specifically to other molecules, allowing them to act as catalysts, structural supports, tiny motors, and so on. The examples we review here by no means exhaust the vast functional repertoire of proteins. However, the specialized functions of the proteins you will encounter elsewhere in this book are based on the same principles.
Question 4–4
Hair is composed largely of fibers of the protein keratin. Individual keratin fibers are covalently cross-linked to one another by many disulfide (S–S) bonds. If curly hair is treated with mild reducing agents that break a few of the cross-links, pulled straight, and then oxidized again, it remains straight. Draw a diagram that illustrates the three different stages of this chemical and mechanical process at the level of the keratin filaments, focusing on the disulfide bonds. What do you think would happen if hair were treated with strong reducing agents that break all the disulfide bonds?
The biological properties of a protein molecule depend on its physical interaction with other molecules. Antibodies attach to viruses or bacteria as part of the body’s defenses; the enzyme hexokinase binds glucose and ATP to catalyze a reaction between them; actin molecules bind to one another to assemble into long filaments; and so on. Indeed, all proteins stick, or bind, to other molecules in a specific manner. In some cases, this binding is very tight; in others, it is weak and short-lived.
The binding of a protein to other biological molecules always shows great specificity: each protein molecule can bind to just one or a few molecules out of the many thousands of different molecules it encounters. Any substance that is bound by a protein—whether it is an ion, a small organic molecule, or a macromolecule—is referred to as a ligand for that protein (from the Latin ligare, “to bind”).
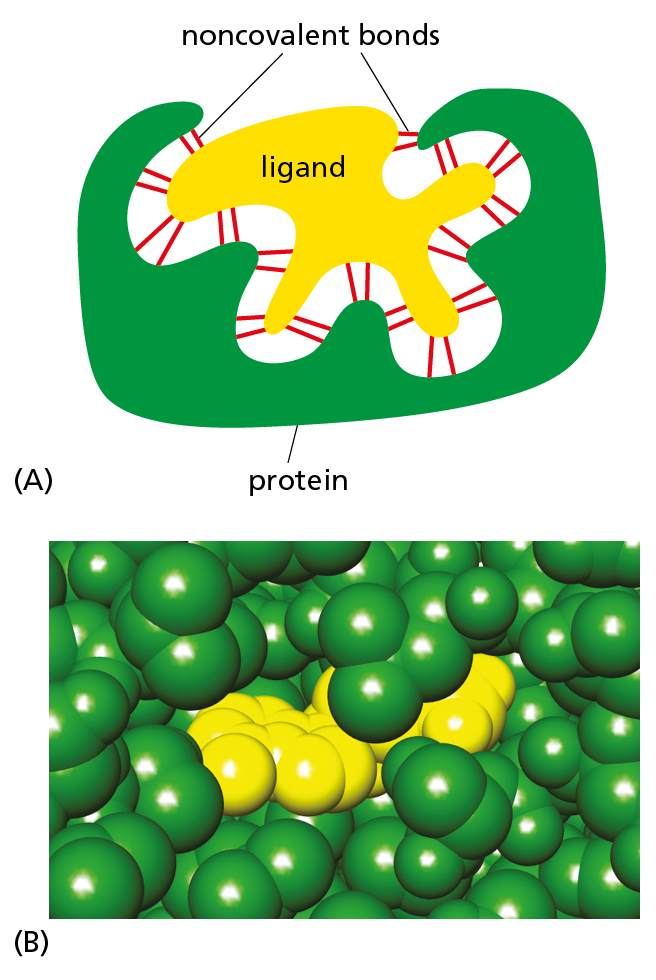
Figure 4–31The binding of a protein to another molecule is highly selective. Many weak interactions are needed to enable a protein to bind tightly to a second molecule (a ligand). The ligand must therefore fit precisely into the protein’s binding site, so that a large number of noncovalent interactions can be formed between the protein and the ligand. (A) Schematic drawing showing the binding of a hypothetical protein and ligand; (B) space-filling model of the ligand–protein interaction shown in Figure 4–32.
The ability of a protein to bind selectively and with high affinity to a ligand is due to the formation of a set of weak, noncovalent interactions—hydrogen bonds, electrostatic attractions, and van der Waals attractions—plus favorable hydrophobic forces (see Panel 2−3, pp. 70–71). Each individual noncovalent interaction is weak, so that effective binding requires many such bonds to be formed simultaneously. This is possible only if the surface contours of the ligand molecule fit very closely to the protein, matching it like a hand in a glove (Figure 4−31).
When molecules have poorly matching surfaces, few noncovalent interactions occur, and the two molecules dissociate as rapidly as they come together. This is what prevents incorrect and unwanted associations from forming between mismatched molecules. At the other extreme, when many noncovalent interactions are formed, the association will persist (see Movie 2.4). Strong binding between molecules occurs in cells whenever a biological function requires that the molecules remain tightly associated for a long time—for example, when a group of macromolecules come together to form a functional subcellular structure such as a ribosome.
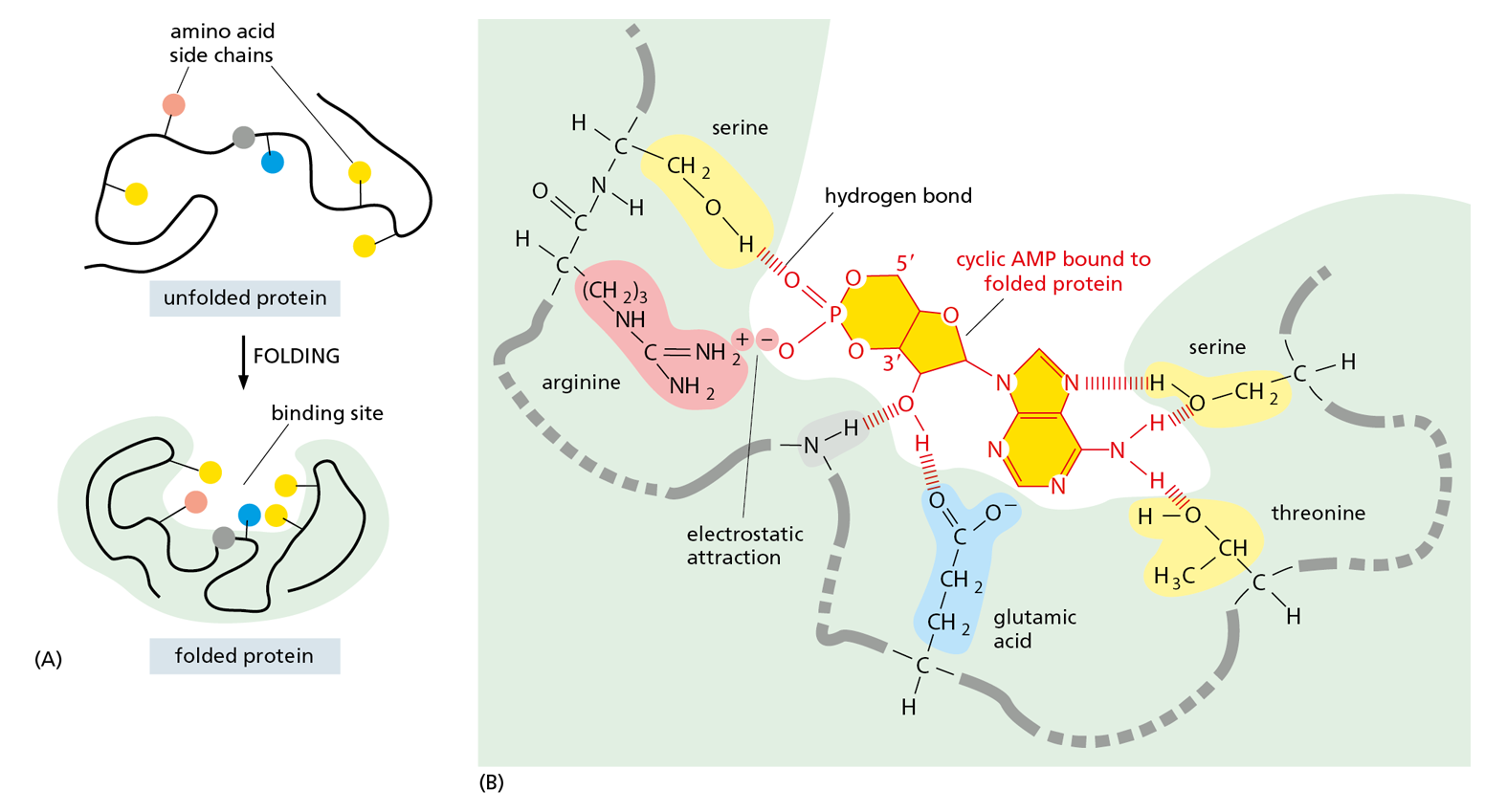
The region of a protein that associates with a ligand, known as its binding site, usually consists of a cavity in the protein surface formed by a particular arrangement of amino acid side chains. These side chains can belong to amino acids that are widely separated on the linear polypeptide chain, but are brought together when the protein folds (Figure 4−32). Other regions on the surface often provide binding sites for different ligands that regulate the protein’s activity, as we discuss later. Still other parts of the protein may be required to attract or attach the protein to a particular location in the cell—for example, the hydrophobic α helix of a membrane-spanning protein allows it to be inserted into the lipid bilayer of a cell membrane (see Figure 4−15 and discussed in Chapter 11).
Figure 4−32Binding sites allow proteins to interact with specific ligands. (A) The folding of the polypeptide chain typically creates a crevice or cavity on the folded protein’s surface, where specific amino acid side chains are brought together in such a way that they can form a set of noncovalent bonds only with certain ligands. (B) Close-up view of an actual binding site showing the hydrogen bonds and an electrostatic interaction formed between a protein and its ligand (in this example, the bound ligand is cyclic AMP, shown in dark yellow).
Although the atoms buried in the interior of a protein have no direct contact with the ligand, they provide an essential framework that gives the surface its contours and chemical properties. Even tiny changes to the amino acids in the interior of a protein can change the protein’s three-dimensional shape and destroy its function.
All proteins must bind to specific ligands to carry out their various functions. For antibodies, the universe of possible ligands is limitless and includes molecules found on bacteria, viruses, and other agents of infection. How does the body manage to produce antibodies capable of recognizing and binding tightly to such a diverse collection of ligands?
Antibodies are immunoglobulin proteins produced by the immune system in response to foreign molecules, especially those on the surface of an invading microorganism. Each antibody binds to a particular target molecule extremely tightly, either inactivating the target directly or marking it for destruction. An antibody recognizes its target molecule, called an antigen, with remarkable specificity. And because there are potentially billions of different antigens we might encounter, humans must be able to produce billions of different antibodies—one of which will be specific for almost any antigen imaginable.
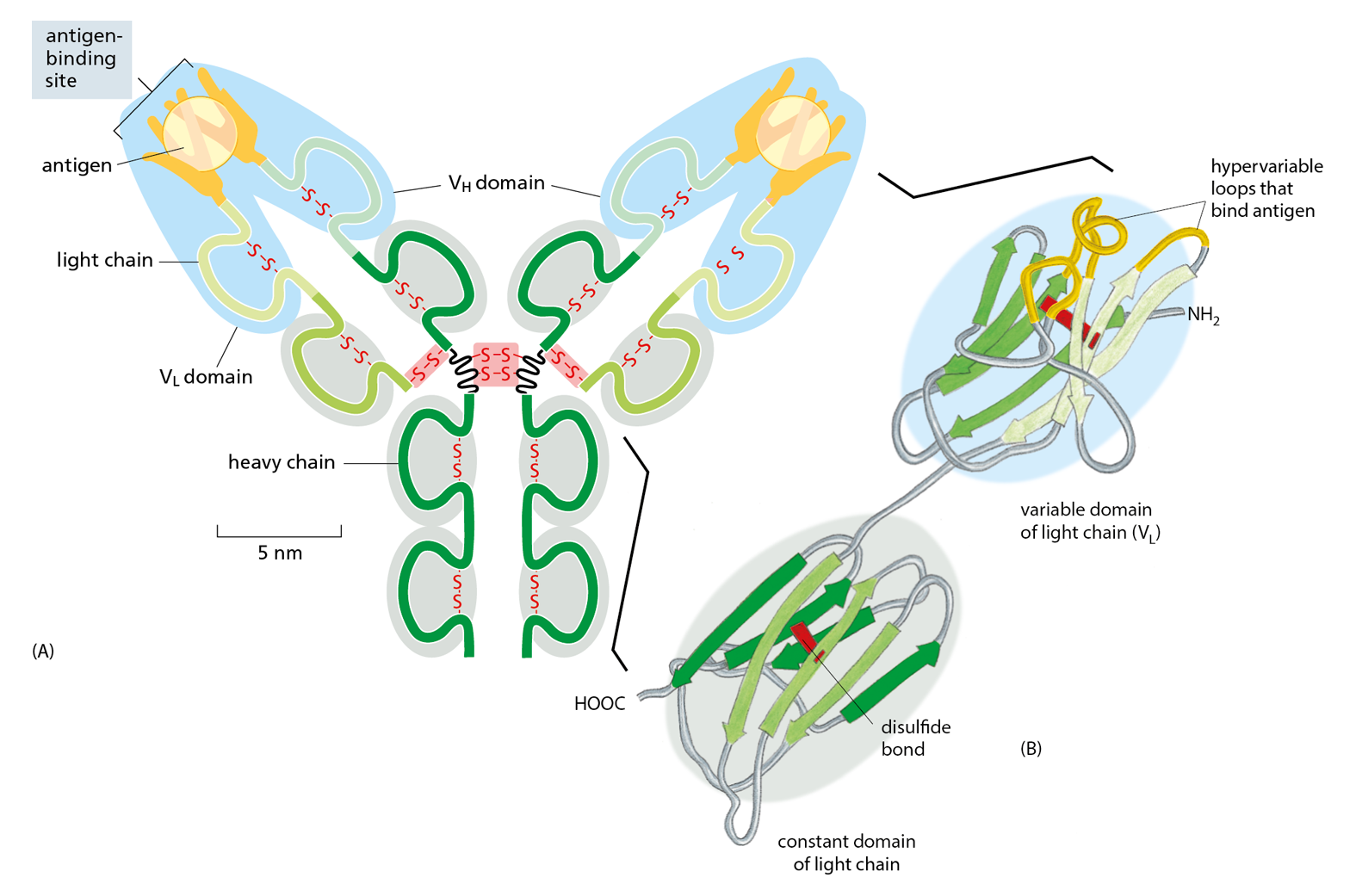
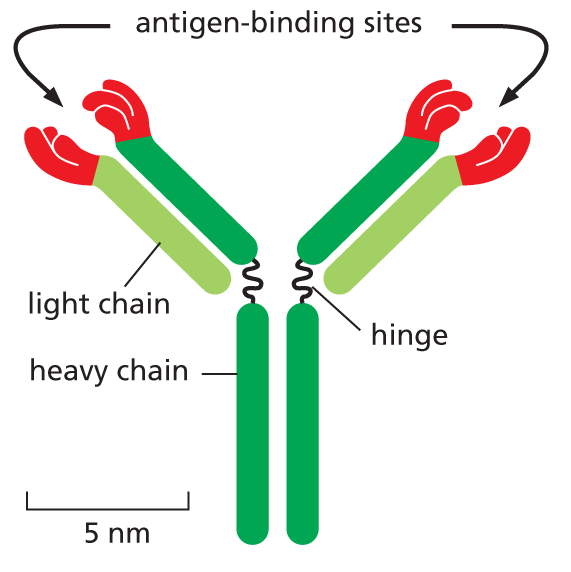
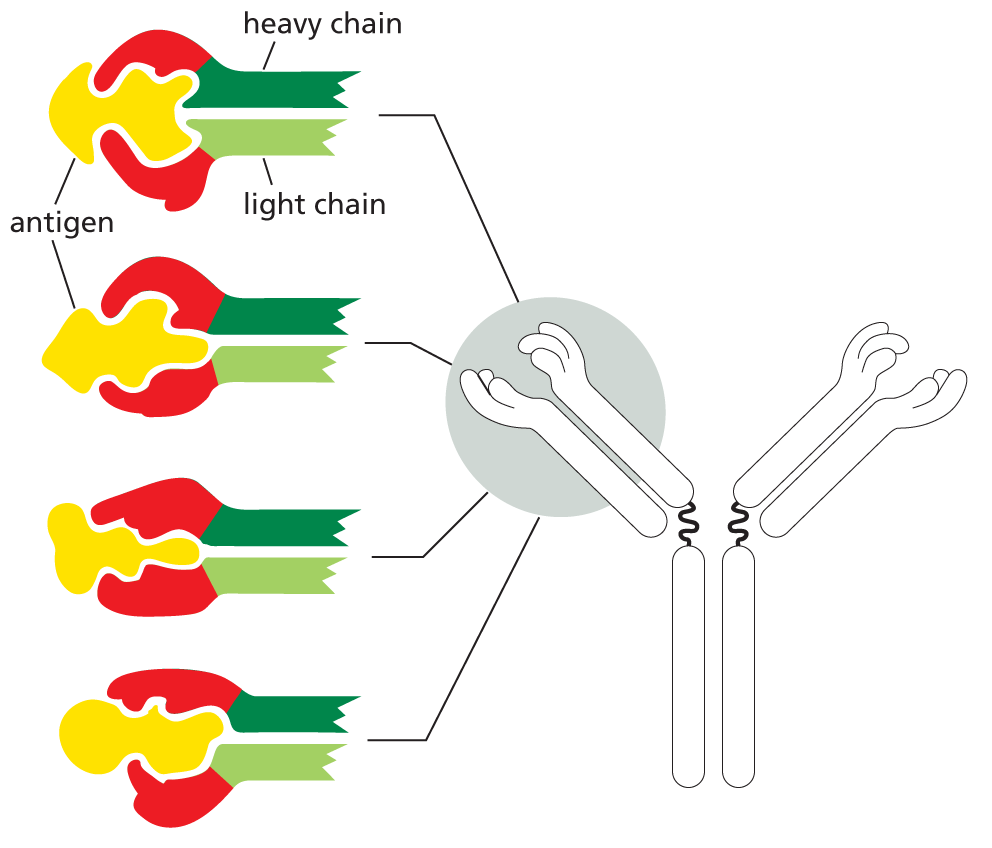
Antibodies are Y-shaped molecules with two identical antigen-binding sites, each of which is complementary to a small portion of the surface of the antigen molecule. A detailed examination of antibody structure reveals that the antigen-binding sites are formed from several loops of polypeptide chain that protrude from the ends of a pair of closely juxtaposed protein domains (Figure 4−33). The amino acid sequence in these loops can vary greatly without altering the basic structure of the antibody. An enormous diversity of antigen-binding sites can therefore be generated by changing only the length and amino acid sequence of these “hypervariable loops,” which is how the wide variety of different antibodies is formed (Movie 4.7).
Figure 4−33An antibody is Y-shaped and has two identical antigen-binding sites, one on each arm of the Y. (A) Schematic drawing of a typical antibody molecule. The protein is composed of four polypeptide chains (two identical heavy chains and two identical, smaller light chains), stabilized and held together by disulfide bonds (red). Each chain is made up of several similar domains, here shaded with blue, for the variable domains, or gray, for the constant domains. The antigen-binding site is formed where a heavy-chain variable domain (VH) and a light-chain variable domain (VL) come close together. These are the domains that differ most in their amino acid sequence in different antibodies—hence their name. (B) Ribbon drawing of a single light chain showing that the most variable parts of the polypeptide chain (orange) extend as loops at one end of the variable domain (VL) to form half of one antigen-binding site of the antibody molecule shown in (A). Note that both the constant and variable domains are composed of a sandwich of two antiparallel β sheets connected by a disulfide bond (red).
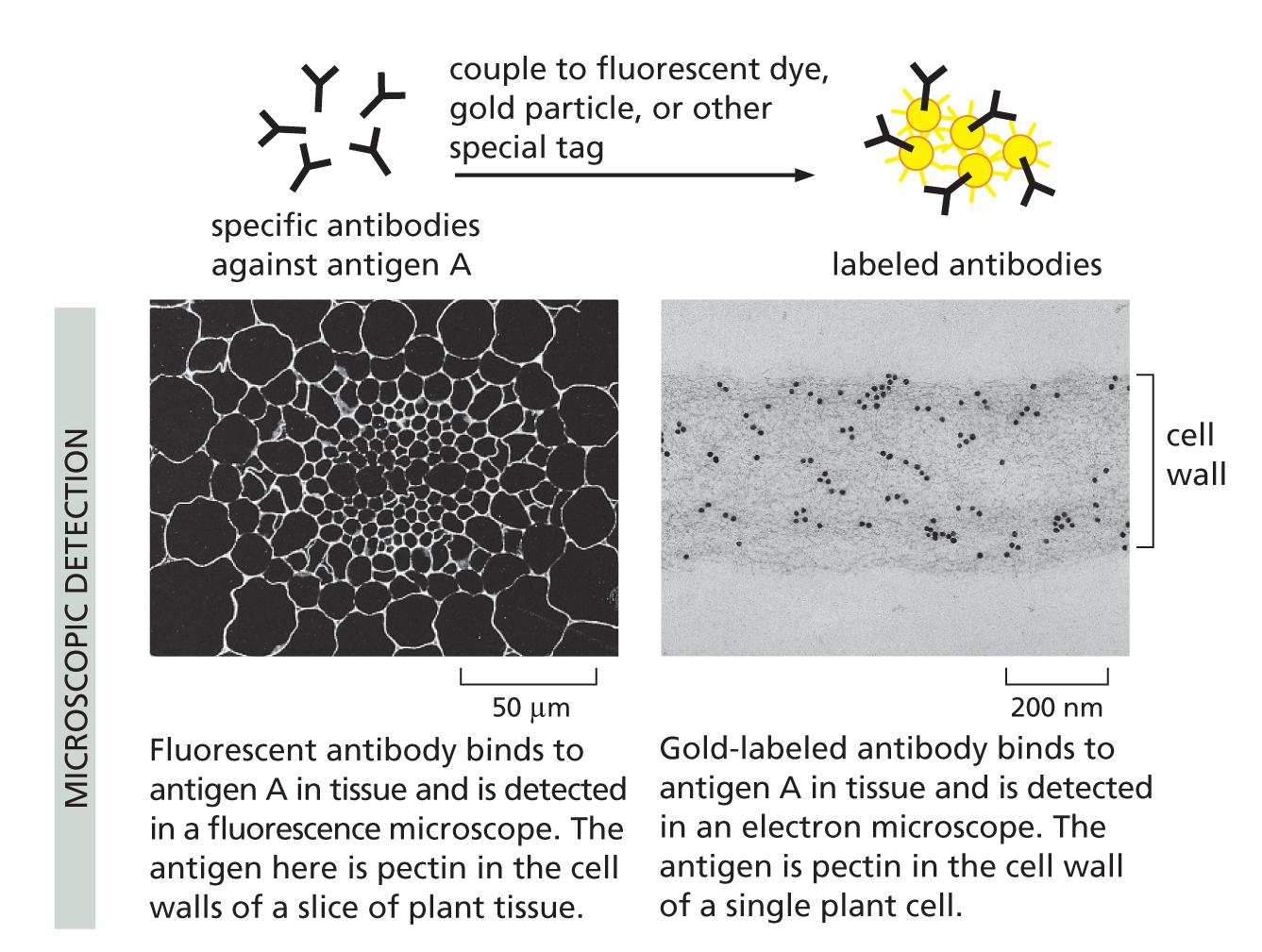
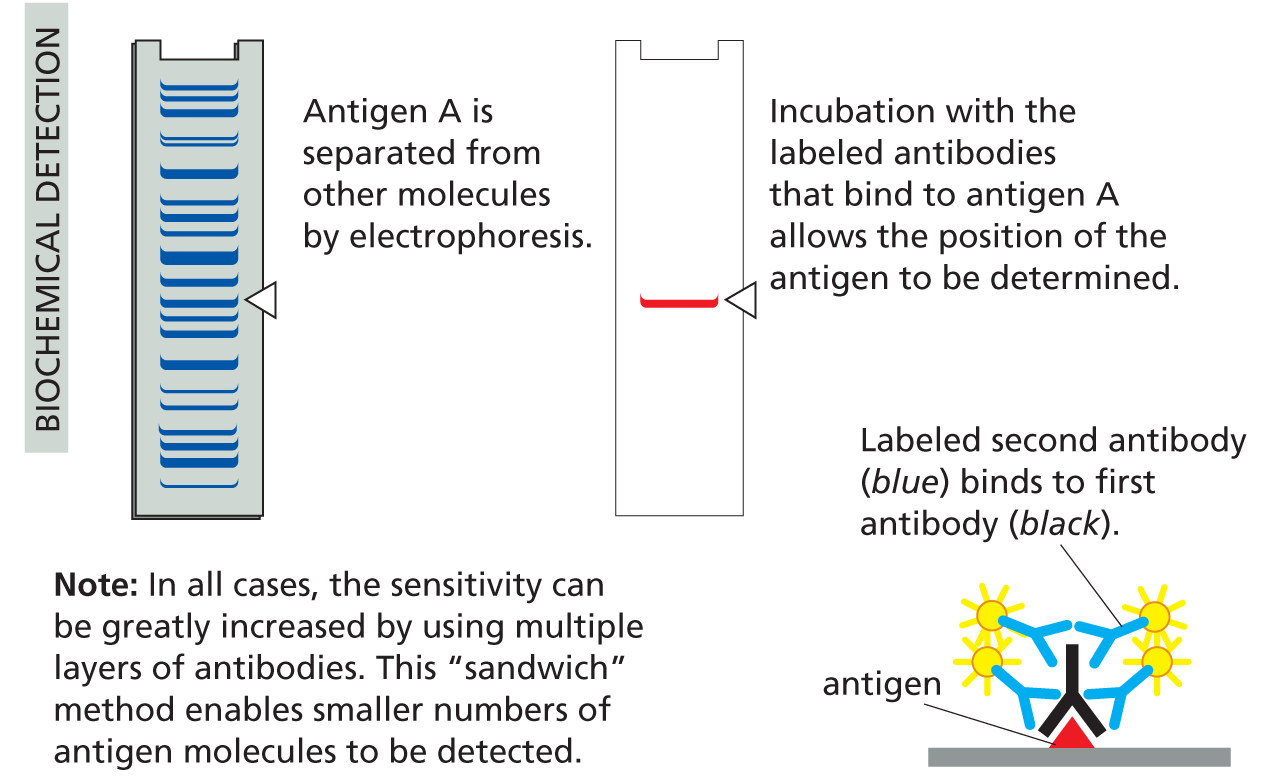
With their unique combination of specificity and diversity, antibodies are not only indispensable for fighting off infections, they are also invaluable in the laboratory, where they can be used to identify, purify, and study other molecules (Panel 4−2, pp. 140–141).
Panel 4–2
making and using antibodies
THE ANTIBODY MOLECULE
Antibodies are proteins that bind very tightly to their targets (antigens). They are produced in vertebrates as a defense against infection. Each antibody molecule is made of two identical light chains and two identical heavy chains. Its two antigen-binding sites are therefore identical. (See Figure 4–33).
ANTIBODY SPECIFICITY
An individual human can make billions of different antibody molecules, each with a distinct antigen-binding site. Each antibody recognizes its antigen with great specificity.
ANTIBODIES DEFEND US AGAINST INFECTION
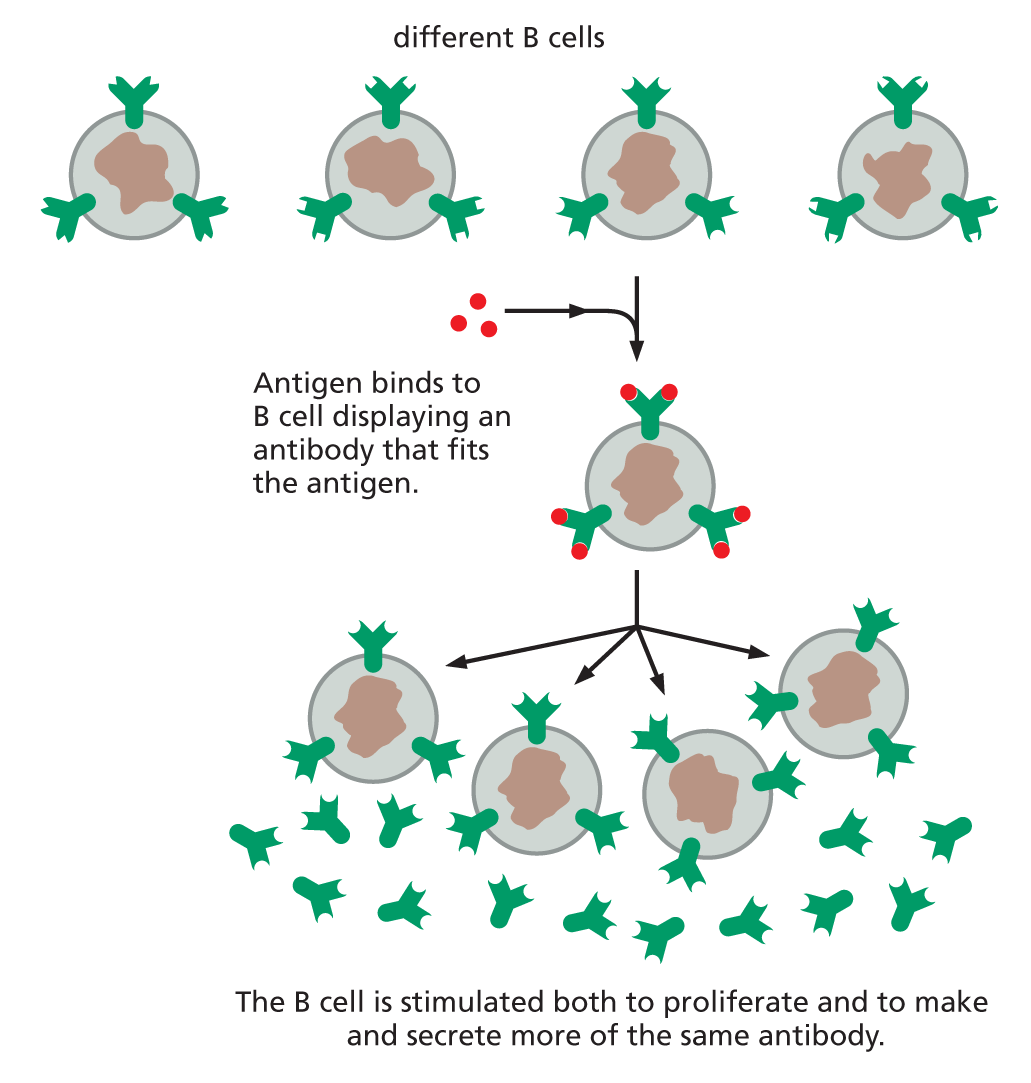
B CELLS PRODUCE ANTIBODIES
Antibodies are made by a class of white blood cells called B lymphocytes, or B cells. Each resting B cell carries a different membrane-bound antibody molecule on its surface that serves as a receptor for recognizing a specific antigen. When antigen binds to this receptor, the B cell is stimulated to divide and to secrete large amounts of the same antibody in a soluble form.
RAISING ANTIBODIES IN ANIMALS
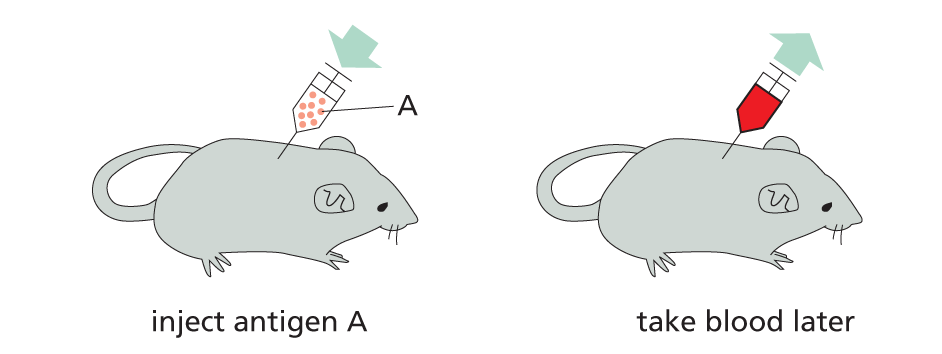
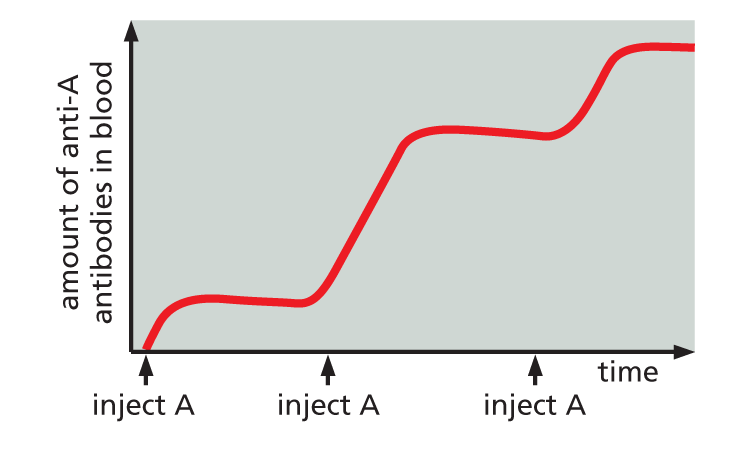
Antibodies can be made in the laboratory by injecting an animal (usually a mouse, rabbit, sheep, or goat) with antigen A.
Repeated injections of the same antigen at intervals of several weeks stimulate specific B cells to secrete large amounts of anti-A antibodies into the bloodstream.
Because many different B cells are stimulated by antigen A, the blood will contain a variety of anti-A antibodies, each of which binds A in a slightly different way.
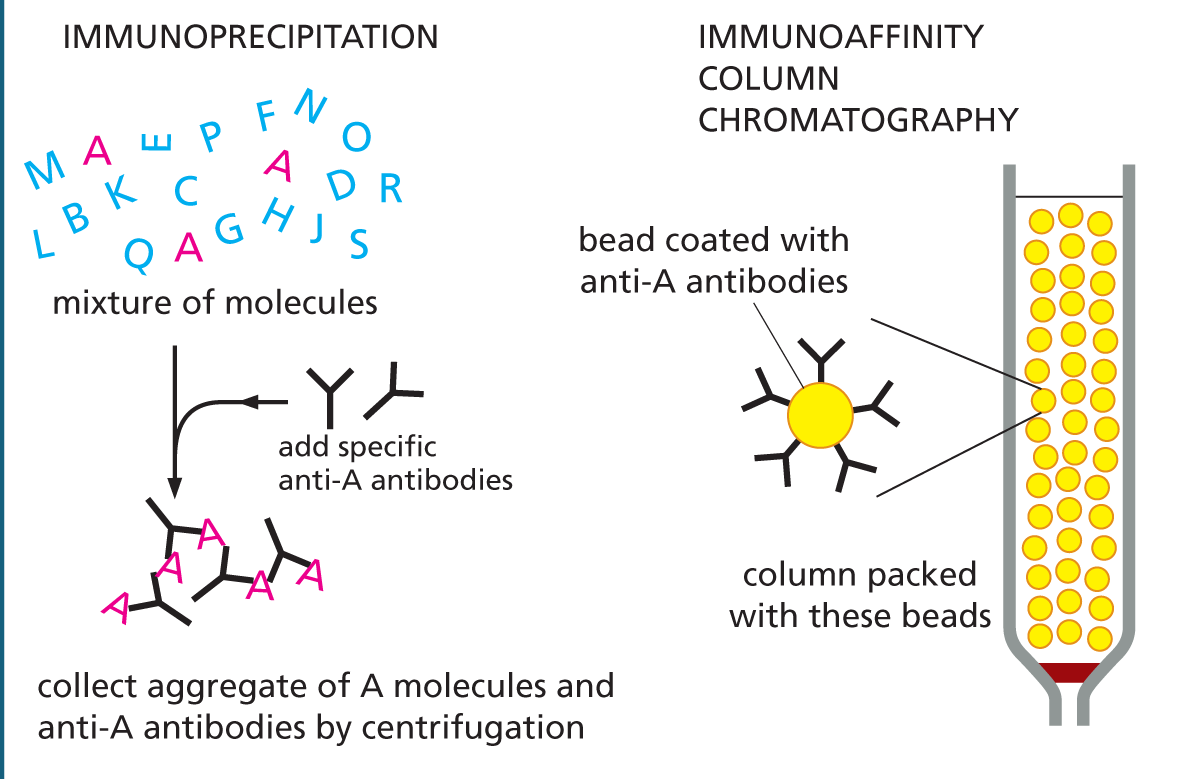
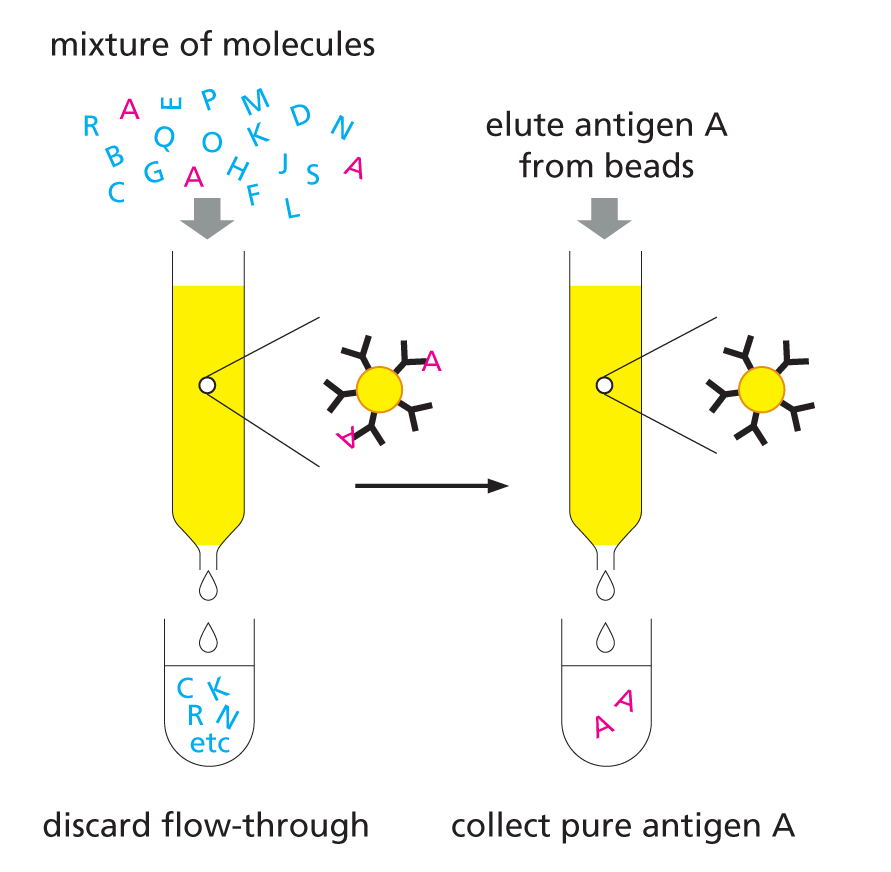
USING ANTIBODIES TO PURIFY MOLECULES
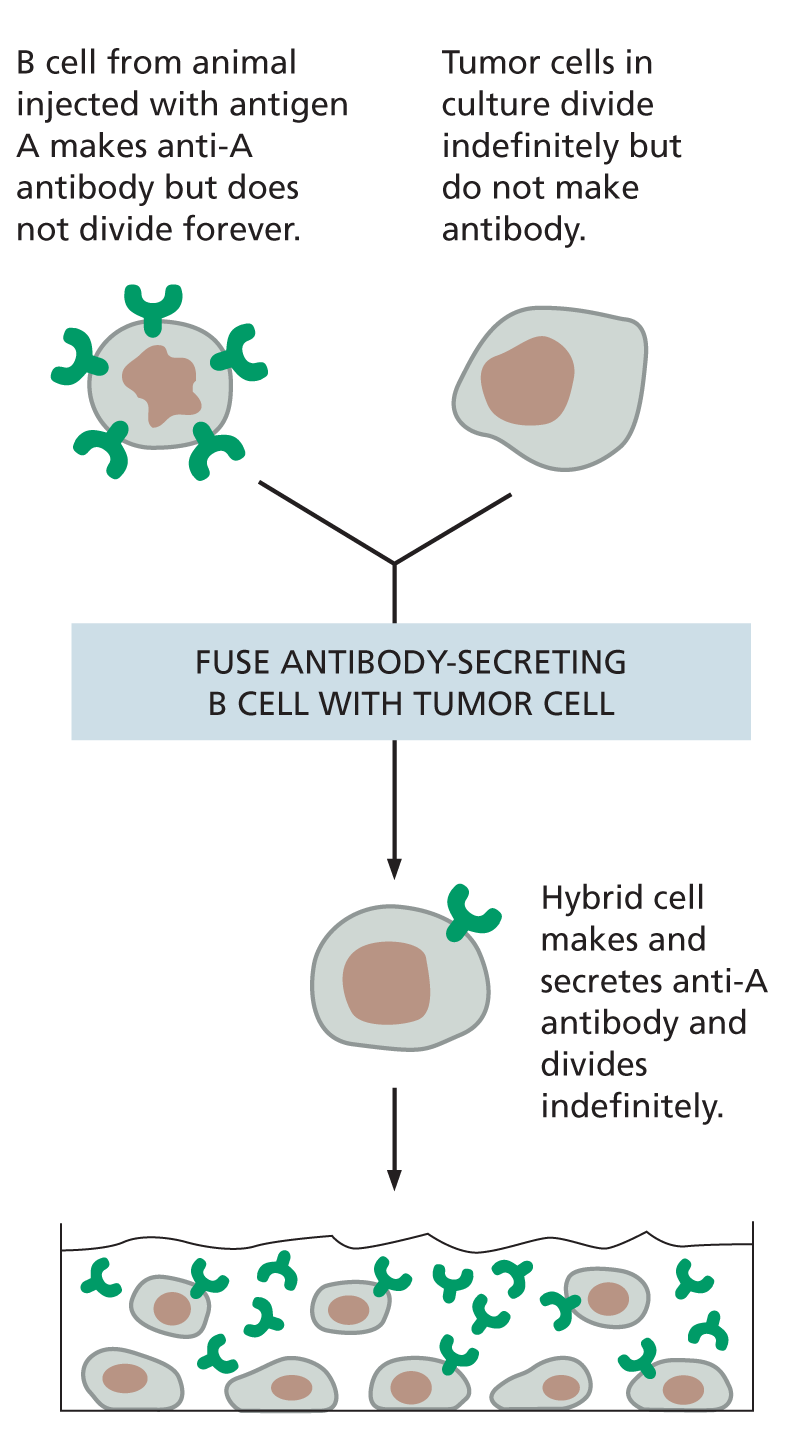
MONOCLONAL ANTIBODIES
Large quantities of a single type of antibody molecule can be obtained by fusing a B cell (taken from an animal injected with antigen A) with a tumor cell. The resulting hybrid cell divides indefinitely and secretes anti-A antibodies of a single (monoclonal) type.
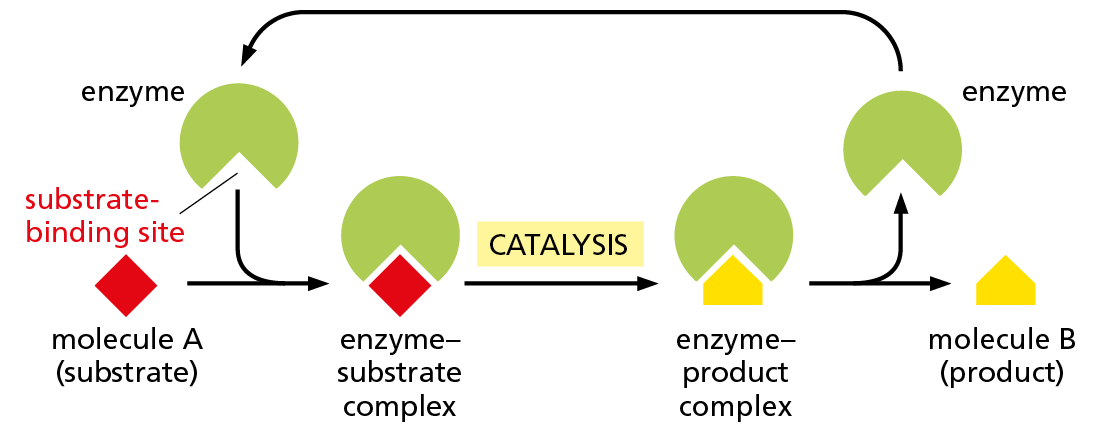
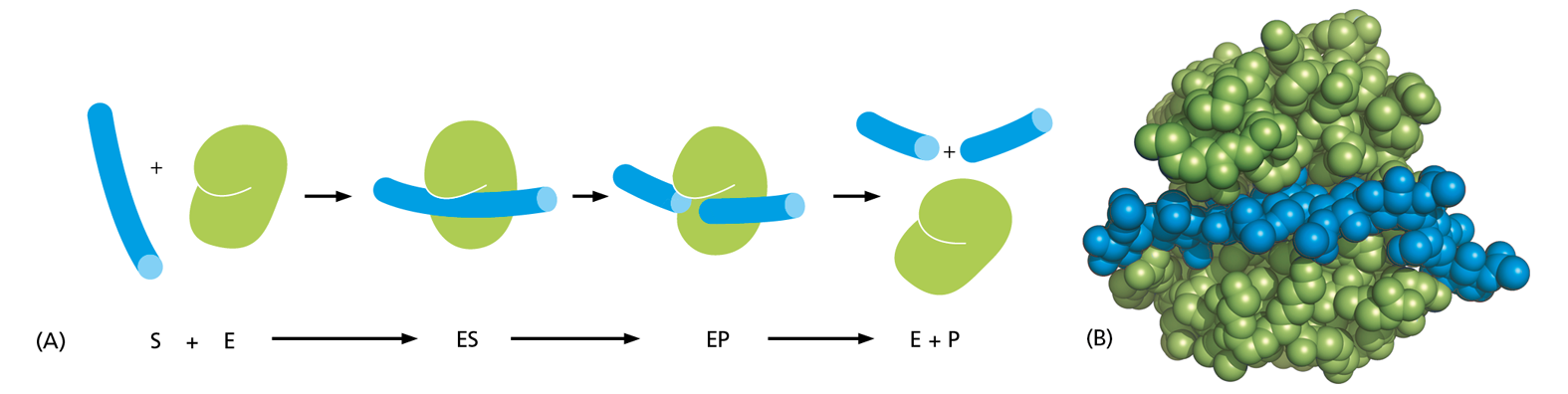
For many proteins, binding to another molecule is their main function. An actin molecule, for example, need only associate with other actin molecules to form a filament. There are proteins, however, for which ligand binding is simply a necessary first step in their function. This is the case for the large and very important class of proteins called enzymes. These remarkable molecules are responsible for nearly all of the chemical transformations that occur in cells. Enzymes bind to one or more ligands, called substrates, and convert them into chemically modified products, doing this over and over again without themselves being changed (Figure 4−34). Thus, enzymes act as catalysts that permit cells to make or break covalent bonds at will. This catalysis of organized sets of chemical reactions by enzymes creates and maintains all cell components, making life possible.
Figure 4−34Enzymes convert substrates to products while remaining unchanged themselves. Each enzyme has a site to which substrate molecules bind, forming an enzyme–substrate complex. There, a covalent bond making and/or breaking reaction occurs, generating an enzyme–product complex. The product is then released, allowing the enzyme to bind additional substrate molecules and repeat the reaction. An enzyme thus serves as a catalyst, and it usually forms or breaks a single covalent bond in a substrate molecule.
Enzymes can be grouped into functional classes based on the chemical reactions they catalyze (Table 4−1). Each type of enzyme is highly specific, catalyzing only a single type of reaction. Thus, hexokinase adds a phosphate group to D-glucose but not to its optical isomer L-glucose; the blood-clotting enzyme thrombin cuts one type of blood-clotting protein between a particular arginine and its adjacent glycine and nowhere else. As discussed in detail in Chapter 3, enzymes often work in sets, with the product of one enzyme becoming the substrate for the next. The result is an elaborate network of metabolic pathways that provides the cell with energy and generates the many large and small molecules that the cell needs.
Table 4–1 some common funCtional classes of enzymes
Enzyme Class
Biochemical Function
Hydrolase
General term for enzymes that catalyze a hydrolytic cleavage reaction
Nuclease
Breaks down nucleic acids by hydrolyzing bonds between nucleotides
Protease
Breaks down proteins by hydrolyzing peptide bonds between amino acids
Ligase
Joins two molecules together; DNA ligase joins two DNA strands together end-to-end
Isomerase
Catalyzes the rearrangement of bonds within a single molecule
Polymerase
Catalyzes polymerization reactions such as the synthesis of DNA and RNA
Kinase
Catalyzes the addition of phosphate groups to molecules. Protein kinases are an important group of kinases that attach phosphate groups to proteins
Phosphatase
Catalyzes the hydrolytic removal of a phosphate group from a molecule
Oxido-reductase
General name for enzymes that catalyze reactions in which one molecule is oxidized while the other is reduced. Enzymes of this type are often called oxidases, reductases, or dehydrogenases
ATPase
Hydrolyzes ATP. Many proteins have an energy-harnessing ATPase activity as part of their function, including motor proteins such as myosin (discussed in Chapter 17) and membrane transport proteins such as the Na+ pump (discussed in Chapter 12)
Enzyme names typically end in “-ase,” with the exception of some enzymes, such as pepsin, trypsin, thrombin, lysozyme, and so on, which were discovered and named before the convention became generally accepted, at the end of the nineteenth century. The name of an enzyme usually indicates the nature of the reaction catalyzed. For example, citrate synthase catalyzes the synthesis of citrate by a reaction between acetyl CoA and oxaloacetate.
The affinities of enzymes for their substrates, and the rates at which they convert bound substrate to product, vary widely from one enzyme to another. Both values can be determined experimentally by mixing purified enzymes and substrates together in a test tube. At a low concentration of substrate, the amount of enzyme−substrate complex—and the rate at which product is formed—will depend solely on the concentration of the substrate. If the concentration of substrate added is large enough, however, all of the enzyme molecules will be filled with substrate. When this happens, the rate of product formation depends on how rapidly the substrate molecule can undergo the reaction that will convert it to product. At this point, the enzymes are working as fast as they can, a value termed Vmax. For many enzymes operating at Vmax, the number of substrate molecules converted to product is in the vicinity of 1000 per second, although turnover numbers ranging from 1 to 100,000 molecules per second have been measured for different enzymes. Enzymes can speed up the rate of a chemical reaction by a factor of a million or more.
Question 4–5
Use drawings to explain how an enzyme (such as hexokinase, mentioned in the text) can distinguish its normal substrate (here, D-glucose) from the optical isomer L-glucose, which is not a substrate. (Hint: remembering that a carbon atom forms four single bonds that are tetrahedrally arranged and that the optical isomers are mirror images of each other around such a bond, draw the substrate as a simple tetrahedron with four different corners and then draw its mirror image. Using this drawing, indicate why only one optical isomer might bind to a schematic active site of an enzyme.)
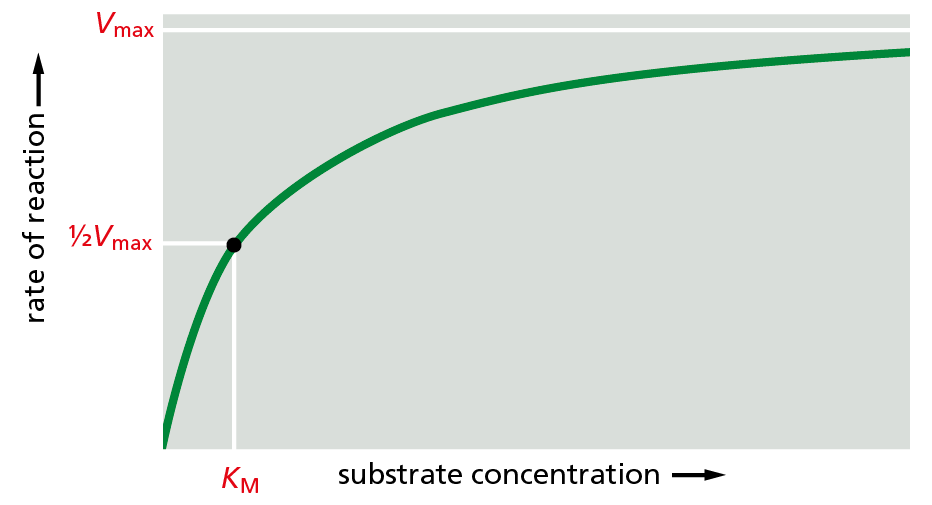
The same type of experiment can be used to gauge how tightly an enzyme interacts with its substrate, a value that is related to how much substrate it takes to fully saturate a sample of enzyme. Because it is difficult to determine at what point an enzyme sample is “fully occupied,” biochemists instead determine the concentration of substrate at which an enzyme works at half its maximum speed. This value, called the Michaelis constant,KM, was named after one of the biochemists who worked out the relationship (Figure 4−35). In general, a small KM indicates that a substrate binds very tightly to the enzyme—due to a large number of noncovalent interactions (see Figure 4−31A); a large KM, on the other hand, indicates weak binding. We describe the methods used to analyze enzyme performance in How We Know, pp. 144–145.
Figure 4−35An enzyme’s performance depends on how rapidly it can process its substrate. The rate of an enzyme reaction (V) increases as the substrate concentration increases, until a maximum value (Vmax) is reached. At this point, all substrate-binding sites on the enzyme molecules are fully occupied, and the rate of the reaction is limited by the rate of the catalytic process on the enzyme surface. For most enzymes, the concentration of substrate at which the reaction rate is half-maximal (KM) is a direct measure of how tightly the substrate is bound, with a large value of KM (a large amount of substrate needed) corresponding to weak binding.
We have discussed how enzymes recognize their substrates. But how do they catalyze the chemical conversion of these substrates into products? To find out, we take a closer look at lysozyme—an enzyme that acts as a natural antibiotic in egg white, saliva, tears, and other secretions. Lysozyme severs the polysaccharide chains that form the cell walls of bacteria. Because the bacterial cell is under pressure due to intracellular osmotic forces, cutting even a small number of polysaccharide chains causes the cell wall to rupture and the bacterium to burst, or lyse—hence the enzyme’s name. Because lysozyme is a relatively small and stable protein, and can be isolated easily in large quantities, it has been studied intensively. It was the first enzyme to have its structure worked out at the atomic level by x-ray crystallography, and its mechanism of action is understood in great detail.
The reaction catalyzed by lysozyme is a hydrolysis: the enzyme adds a molecule of water to a single bond between two adjacent sugar groups in the polysaccharide chain, thereby causing the bond to break (see Figure 2−19). This reaction is energetically favorable because the free energy of the severed polysaccharide chains is lower than the free energy of the intact chain. However, the pure polysaccharide can sit for years in water without being hydrolyzed to any detectable degree. This is because there is an energy barrier to such reactions, called the activation energy (discussed in Chapter 3, pp. 89–90). For a colliding water molecule to break a bond linking two sugars, the polysaccharide molecule has to be distorted into a particular shape—the transition state—in which the atoms around the bond have an altered geometry and electron distribution. To distort the polysaccharide in this way requires a large input of energy—which is where the enzyme comes in.
How We Know
MEASURING ENZYME PERFORMANCE
At first glance, it seems that a cell’s metabolic pathways have been pretty well mapped out, with each reaction proceeding predictably to the next. So why would anyone need to know exactly how tightly a particular enzyme clutches its substrate or whether it can process 100 or 1000 substrate molecules every second?
In reality, metabolic maps merely suggest which pathways a cell might follow as it converts nutrients into small molecules, chemical energy, and the larger building blocks of life. Like a road map, they do not predict the density of traffic under a particular set of conditions; that is, which pathways the cell will use when it is starving, when it is well fed, when oxygen is scarce, when it is stressed, or when it decides to divide. The study of an enzyme’s kinetics—how fast it operates, how it handles its substrate, how its activity is controlled—allows us to predict how an individual catalyst will perform, and how it will interact with other enzymes in a network. Such knowledge leads to a deeper understanding of cell biology, and it opens the door to learning how to harness enzymes to perform desired reactions, including the large-scale production of specific chemicals.
Speed
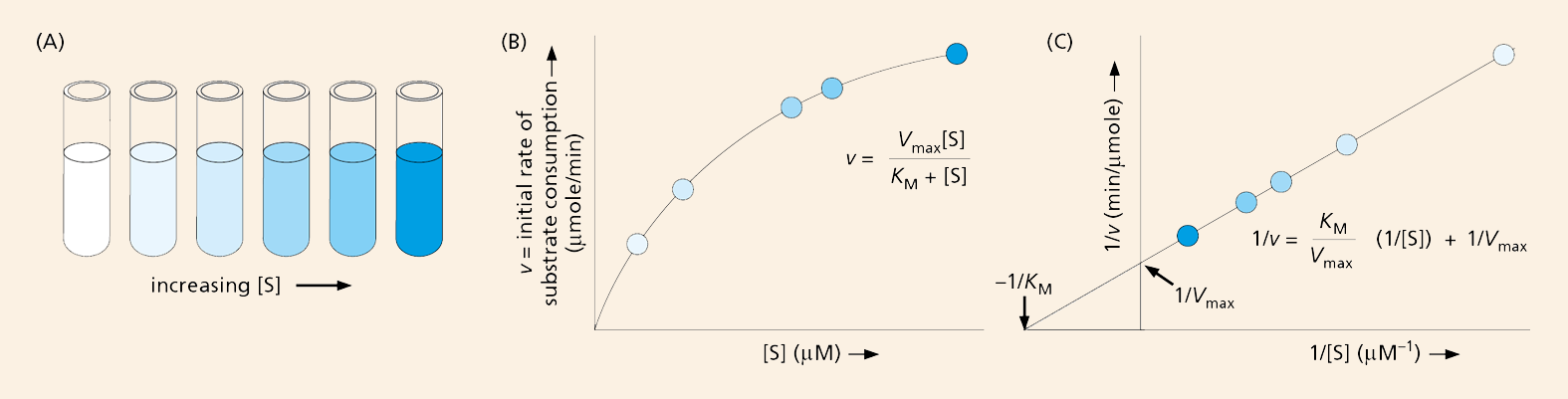
The first step to understanding how an enzyme performs involves determining the maximal velocity, Vmax, for the reaction it catalyzes. This is accomplished by measuring, in a test tube, how rapidly the reaction proceeds in the presence of a fixed amount of enzyme and different concentrations of substrate (Figure 4–36A): the rate should increase as the amount of substrate rises until the reaction reaches its Vmax (Figure 4–36B). The velocity of the reaction can be measured by monitoring either how quickly the substrate is consumed or how rapidly the product accumulates. In many cases, the appearance of product or the disappearance of substrate can be observed directly with a spectrophotometer. This instrument detects the presence of molecules that absorb light at a particular wavelength; NADH, for example, absorbs light at 340 nm, while its oxidized counterpart, NAD+, does not. So, a reaction that generates NADH (by reducing NAD+) can be monitored by following the formation of NADH at 340 nm in a spectrophotometer.
Figure 4–36Measured reaction rates are plotted to determine theVmaxandKMof an enzyme-catalyzed reaction. (A) Test tubes containing a series of increasing substrate concentrations are prepared, a fixed amount of enzyme is added, and initial reaction rates (velocities) are determined. (B) The initial velocities (v) plotted against the substrate concentrations [S] give a curve described by the general equation y = ax/(b + x). Substituting our kinetic terms, the equation becomes v = Vmax[S]/(KM + [S]), where Vmax is the asymptote of the curve (the value of y at an infinite value of x), and KM is equal to the substrate concentration where v is one-half Vmax. This is called the Michaelis–Menten equation, named for the biochemists who provided evidence for this enzymatic relationship. (C) In a double-reciprocal plot, 1/v is plotted against 1/[S]. The equation describing this straight line is 1/v = (KM/Vmax)(1/[S]) + 1/Vmax. When 1/[S] = 0, the y intercept (1/v) is 1/Vmax. When 1/v = 0, the x intercept (1/[S]) is –1/KM. Plotting the data this way allows Vmax and KM to be calculated more precisely. By convention, lowercase letters are used for variables (hence v for velocity) and uppercase letters are used for constants (hence Vmax).
Looking at the plot in Figure 4–36B, however, it is difficult to determine the exact value of Vmax, as it is not clear where the reaction rate will reach its plateau. To get around this problem, the data are converted to their reciprocals and graphed in a “double-reciprocal plot,” where the inverse of the velocity (1/v) appears on the y axis and the inverse of the substrate concentration (1/[S]) on the x axis (Figure 4–36C). This graph yields a straight line whose y intercept (the point where the line crosses the y axis) represents 1/Vmax and whose x intercept corresponds to –1/KM. These values are then converted to values for Vmax and KM.
Control
Substrates are not the only molecules that can influence how well or how quickly an enzyme works. In many cases, products, substrate lookalikes, inhibitors, and other small molecules can also increase or decrease enzyme activity. Such regulation allows cells to control when and how rapidly various reactions occur, a process we discuss in detail in this chapter.
The effect of an inhibitor on an enzyme’s activity is monitored in the same way that we measured the enzyme’s kinetics. A curve is first generated showing the velocity of the uninhibited reaction between enzyme and substrate. Additional curves are then produced for reactions in which the inhibitor molecule has been included in the mix.
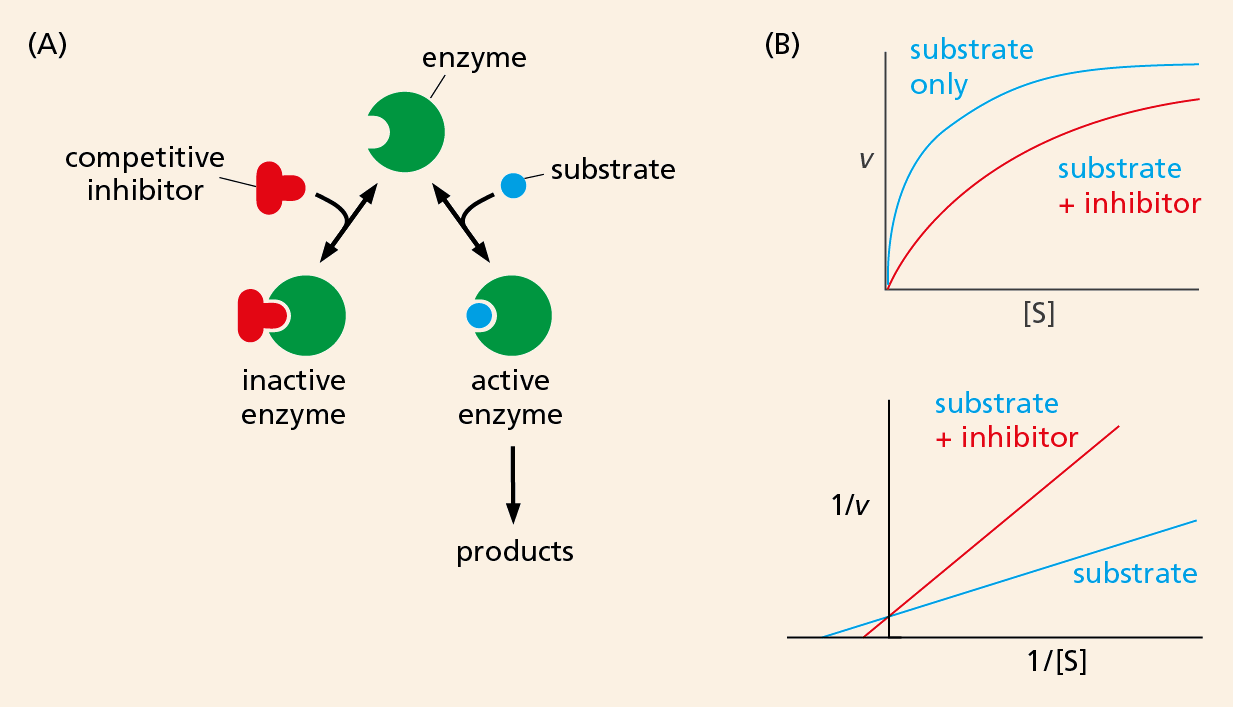
Comparing these curves, with and without inhibitor, can also reveal how a particular inhibitor impedes enzyme activity. For example, some inhibitors bind to the same site on an enzyme as its substrate. These competitive inhibitors block enzyme activity by competing directly with the substrate for the enzyme’s attention. They resemble the substrate enough to tie up the enzyme, but they differ enough in structure to avoid getting converted to product. This blockage can be overcome by adding enough substrate so that enzymes are more likely to encounter a substrate molecule than an inhibitor molecule. From the kinetic data, we can see that competitive inhibitors do not change the Vmax of a reaction; in other words, add enough substrate and the enzyme will encounter mostly substrate molecules and will reach its maximum velocity (Figure 4–37).
Figure 4–37A competitive inhibitor directly blocks substrate binding to an enzyme. (A) The active site of the enzyme can bind either the competitive inhibitor or the substrate, but not both together. (B) The upper plot shows that inhibition by a competitive inhibitor can be overcome by increasing the substrate concentration. The double-reciprocal plot below shows that the Vmax of the reaction is not changed in the presence of the competitive inhibitor: the y intercept is identical for both the curves.
Competitive inhibitors can be used to treat patients who have been poisoned by ethylene glycol, an ingredient in commercially available antifreeze. Although ethylene glycol is itself not fatally toxic, a by-product of its metabolism—oxalic acid—can be lethal. To prevent oxalic acid from forming, the patient is given a large (though not quite intoxicating) dose of ethanol. Ethanol competes with the ethylene glycol for binding to alcohol dehydrogenase, the first enzyme in the pathway to oxalic acid formation. As a result, the ethylene glycol remains mostly unmetabolized and is safely eliminated from the body.
Other types of inhibitors may interact with sites on the enzyme distant from where the substrate binds. Many biosynthetic enzymes are regulated by feedback inhibition, whereby an enzyme early in a pathway will be shut down by a product generated later in the pathway (see, for example, Figure 4–43). Because this type of inhibitor binds to a separate, regulatory site on the enzyme, the substrate can still bind, but it might do so more slowly than it would in the absence of inhibitor. Such noncompetitive inhibition is not overcome by the addition of more substrate.
Design
With the kinetic data in hand, we can use computer modeling programs to predict how an enzyme will perform, and even how a cell will respond, when exposed to different conditions—such as the addition of a particular sugar or amino acid to the culture medium, or the addition of a poison or a pollutant. Seeing how a cell manages its resources—which pathways it favors for dealing with particular biochemical challenges—can also suggest strategies for designing better catalysts for reactions of medical or commercial importance (e.g., for producing drugs or detoxifying industrial waste). Using such tactics, bacteria have even been genetically engineered to produce large amounts of indigo—the dye, originally extracted from plants, that makes your blue jeans blue. We discuss the methods that enable such genetic manipulation in detail in Chapter 10.
Harnessing the power of cell biology for commercial purposes—even to produce something as simple as the amino acid tryptophan—is currently a multibillion-dollar industry. And, as more genome data come in, presenting us with more enzymes to exploit, vats of custom-made bacteria are increasingly churning out drugs and chemicals that represent the biological equivalent of pure gold.
Like all enzymes, lysozyme has a binding site on its surface, termed an active site, which is where catalysis takes place. Because its substrate is a polymer, lysozyme’s active site is a long groove that cradles six of the linked sugars in the polysaccharide chain at the same time. Once this enzyme–substrate complex forms, the enzyme cuts the polysaccharide by catalyzing the addition of a water molecule to one of its sugar–sugar bonds, and the severed chains are then quickly released, freeing the enzyme for further cycles of cleavage (Figure 4−38).
Figure 4−38Lysozyme cleaves a polysaccharide chain. (A) Schematic view of the enzyme lysozyme (E), which catalyzes the cutting of a polysaccharide substrate molecule (S). The enzyme first binds to the polysaccharide to form an enzyme–substrate complex (ES), then it catalyzes the cleavage of a specific covalent bond in the backbone of the polysaccharide. The resulting enzyme–product complex (EP) rapidly dissociates, releasing the products (P) and leaving the enzyme free to act on another substrate molecule. (B) A space-filling model of lysozyme bound to a short length of polysaccharide chain prior to cleavage.
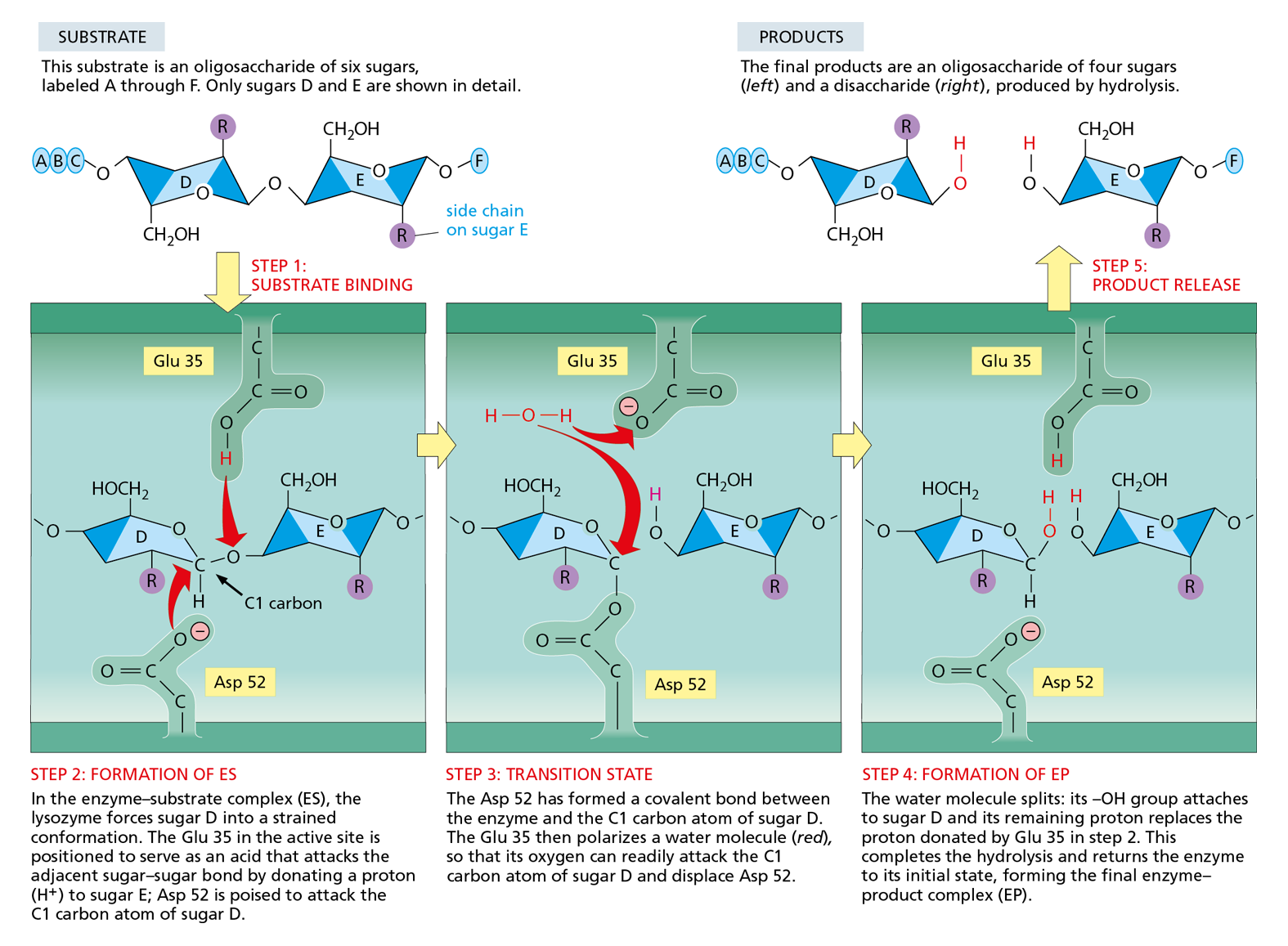
Like any protein binding to its ligand, lysosome recognizes its substrate through the formation of multiple noncovalent bonds (see Figure 4−32). However, lysozyme holds its polysaccharide substrate in such a way that one of the two sugars involved in the bond to be broken is distorted from its normal, most stable conformation. Conditions are thereby created in the microenvironment of the lysozyme active site that greatly reduce the activation energy necessary for the hydrolysis to take place (Figure 4−39). Because the activation energy is so low, the overall chemical reaction—from the initial binding of the polysaccharide to the final release of the severed chains—occurs many millions of times faster in the presence of lysozyme than it would in its absence. In the absence of lysozyme, the energy of random molecular collisions almost never exceeds the activation energy required for the reaction to occur; the hydrolysis of such polysaccharides thus occurs extremely slowly, if at all.
Figure 4−39Enzymes bind to, and chemically alter, substrate molecules. In the active site of lysozyme, a covalent bond in a polysaccharide molecule is bent and then broken. The top row shows the free substrate and the free products. The three lower panels depict sequential events at the enzyme active site, during which a sugar–sugar covalent bond is broken. Note the change in the conformation of sugar D in the enzyme–substrate complex compared with the free substrate. This conformation favors the formation of the transition state shown in the middle panel, greatly lowering the activation energy required for the reaction. The reaction, and the structure of lysozyme bound to its product, are shown in Movie 4.8 and Movie 4.9. (Based on D.J. Vocadlo et al., Nature 412:835–838, 2001.)
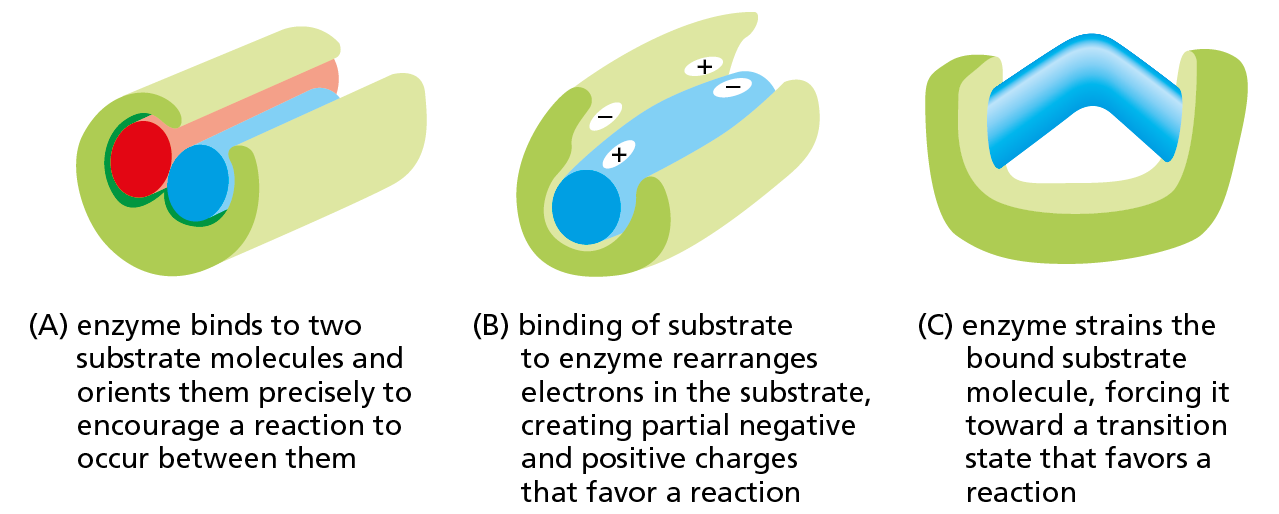
Other enzymes use similar mechanisms to lower the activation energies and speed up the reactions they catalyze. In reactions involving two or more substrates, the active site acts like a template or mold that brings the reactants together in the proper orientation for the reaction to occur (Figure 4−40A). As we saw for lysozyme, the active site can also contain precisely positioned chemical groups that speed up the reaction by altering the distribution of electrons in the substrates (Figure 4−40B). Binding to the enzyme also changes the shape of the substrate, bending bonds so as to drive the bound molecule toward a particular transition state (Figure 4−40C). Finally, like lysozyme, many enzymes participate intimately in the reaction by briefly forming a covalent bond between the substrate and an amino acid side chain in the active site. Subsequent steps in the reaction restore the side chain to its original state, so that the enzyme remains unchanged after the reaction and can go on to catalyze many more reactions.
Figure 4−40Enzymes can encourage a reaction in several ways. (A) Holding reacting substrates together in a precise alignment. (B) Rearranging the distribution of charge in a reaction intermediate. (C) Altering bond angles in the substrate to increase the rate of a particular reaction. A single enzyme may use any of these mechanisms in combination.
Many of the drugs we take to treat or prevent illness work by blocking the activity of a particular enzyme. Cholesterol-lowering statins inhibit HMG-CoA reductase, an enzyme involved in the synthesis of cholesterol by the liver. Methotrexate kills some types of cancer cells by shutting down dihydrofolate reductase, an enzyme that produces a compound required for DNA synthesis during cell division. Because cancer cells have lost important intracellular control systems, some of them are unusually sensitive to treatments that interrupt chromosome replication, making them susceptible to methotrexate.
Pharmaceutical companies often develop drugs by first using automated methods to screen massive libraries of compounds to find chemicals that are able to inhibit the activity of an enzyme of interest. They can then chemically modify the most promising compounds to make them even more effective, enhancing their binding affinity, specificity for the target enzyme, and persistence in the human body. As we discuss in Chapter 20, the anticancer drug Gleevec® was designed to specifically inhibit an enzyme whose aberrant behavior is required for the growth of a type of cancer called chronic myeloid leukemia. The drug binds tightly in the substrate-binding pocket of that enzyme, blocking its activity.
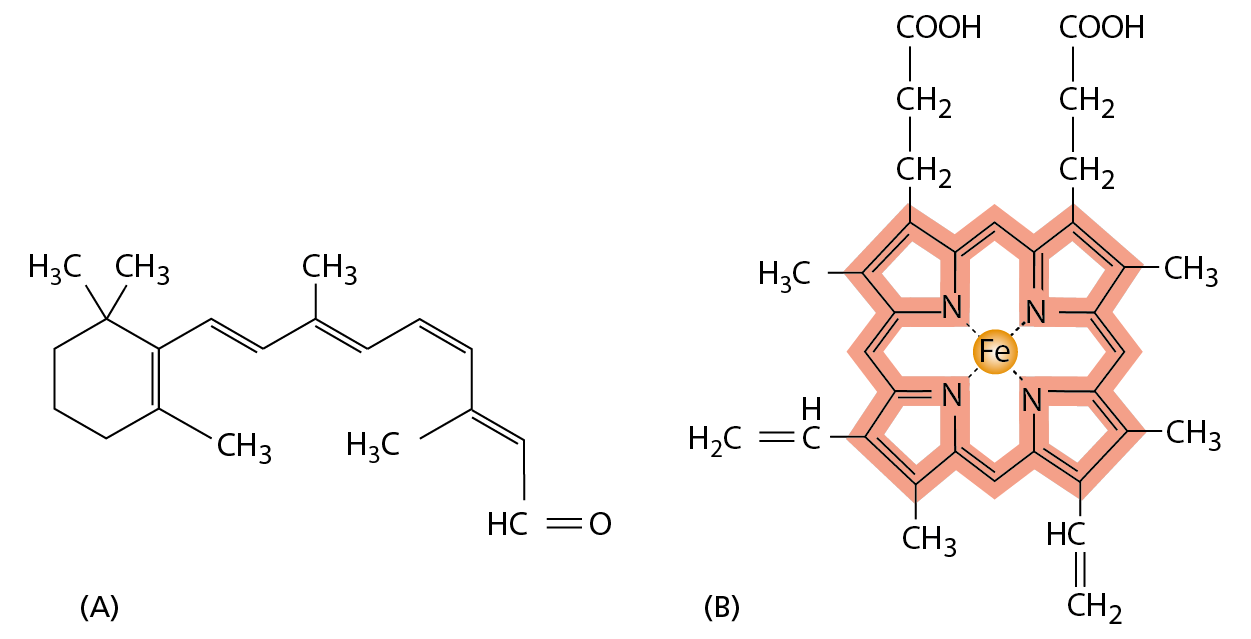
Although the precise order of their amino acids gives proteins their shape and functional versatility, sometimes amino acids by themselves are not enough for a protein to do its job. Just as we use tools to enhance and extend the capabilities of our hands, so proteins often employ small, nonprotein molecules to perform functions that would be difficult or impossible using amino acids alone. Thus, the photoreceptor protein rhodopsin, which is the light-sensitive protein made by the rod cells in the retina of our eyes, detects light by means of a small molecule, retinal, which is attached to the protein by a covalent bond to a lysine side chain (Figure 4−41A). Retinal changes its shape when it absorbs a photon of light, and this change is amplified by rhodopsin to trigger a cascade of reactions that eventually leads to an electrical signal being carried to the brain.
Figure 4−41Retinal and heme are required for the function of certain proteins. (A) The structure of retinal, the light-sensitive molecule covalently attached to the rhodopsin protein in our eyes. (B) The structure of a heme group, shown with the carbon-containing heme ring colored red and the iron atom at its center in orange. A heme group is tightly, but noncovalently, bound to each of the four polypeptide chains in hemoglobin, the oxygen-carrying protein whose structure was shown in Figure 4−24.
Another example of a protein that contains a nonprotein portion essential for its function is hemoglobin (see Figure 4−24). A molecule of hemoglobin carries four noncovalently bound heme groups, ring-shaped molecules each with a single central iron atom (Figure 4−41B). Heme gives hemoglobin—and blood—its red color. By binding reversibly to dissolved oxygen gas through its iron atom, heme enables hemoglobin to pick up oxygen in the lungs and release it in tissues that need it.
Enzymes, too, make use of nonprotein molecules: they frequently have a small molecule or metal atom associated with their active site that assists with their catalytic function. Carboxypeptidase, an enzyme that cuts polypeptide chains, carries a tightly bound zinc ion in its active site. During the cleavage of a peptide bond by carboxypeptidase, the zinc ion forms a transient bond with one of the substrate atoms, thereby assisting the hydrolysis reaction. In other enzymes, a small organic molecule—often referred to as a coenzyme—serves a similar purpose. Biotin, for example, is found in enzymes that transfer a carboxyl group (–COO–) from one molecule to another (see Figure 3−38). Biotin participates in these reactions by forming a covalent bond to the –COO– group to be transferred, thereby producing an activated carrier (see Table 3–2, p. 109). This small molecule is better suited for this function than any of the amino acids used to make proteins.
Because biotin cannot be synthesized by humans, it must be provided in the diet; thus biotin is classified as a vitamin. Other vitamins are similarly needed to make small molecules that are essential components of our proteins; vitamin A, for example, is needed in the diet to make retinal, the light-sensitive part of rhodopsin.
Region on the surface of a protein, typically a cavity or groove, that interacts with another molecule (a ligand) through the formation of multiple noncovalent bonds.
Protein produced by B lymphocytes in response to a foreign molecule or invading organism. Binds to the foreign molecule or cell extremely tightly, thereby inactivating it or marking it for destruction.
Enzyme that severs the polysaccharide chains that form the cell walls of bacteria; found in many secretions including saliva and tears, where it serves as an antibiotic.
Transient structure that forms during the course of a chemical reaction; in this configuration, a molecule has the highest free energy; it is no longer the substrate, but is not yet the product.