Immunological memory and the secondary immune response
In previous chapters we saw how an adaptive immune response is made against any pathogen that outruns the forces of innate immunity and successfully invades a person’s body for the first time. In this circumstance, the infection causes disease before the primary adaptive immune response can clear the infection. Because the pathogen has invaded successfully on one occasion, it is likely to do so again. Anticipating such incursions, the adaptive immune system retains a memory of its battles with pathogens, which enables it to capitalize on past experience when confronted by a reinvading pathogen. This immunological memory allows a person to react to subsequent infections by the pathogen with a secondary immune response that is quicker and stronger than the primary response. In most instances, the secondary response is so effective that the infection is cleared without any significant symptoms of disease. This part of the chapter examines how immunological memory is formed in the course of the primary immune response to a pathogen and is used to make stronger secondary immune responses to subsequent infections by the same pathogen.
11-1Immunological memory is essential for the survival of human populations
The nature of immunological memory and its benefit are well illustrated by Peter Panum’s classic epidemiological study of the inhabitants of the Faroe Islands in the North Atlantic Ocean. Measles virus, a highly infectious and life-threatening pathogen, was first introduced to the islands in 1781, when it caused an epidemic in which the entire human population was infected and suffered disease. More than 60 years later, in 1846, when the measles virus was again brought to the islands, almost all of the 5000 inhabitants who had been born since the first epidemic came down with the disease. But all 98 survivors of the 1781 epidemic proved resistant: they had retained sufficient immunological memory, also called protective immunity, to prevent their second exposure to the measles virus from becoming a disease-causing infection.
Until the latter part of the 20th century, smallpox was, like measles, a much-feared killer of humankind: from 1850 to 1979 about 1 billion people died from smallpox infection. During this same period, worldwide vaccination programs progressively reduced the spread of smallpox virus to a point where mass vaccination was discontinued in the United States in 1972. By 1979 smallpox had been eradicated. A course of two immunizations with vaccinia virus, a close but benign relative of the smallpox virus, is sufficient to induce a secondary immune response with an immunological memory that also works against smallpox. At present, about half the population of the United States has been vaccinated against smallpox and half has not. Because neither group has ever been exposed to smallpox virus, comparison of the two groups has revealed much about the persistence of immunological memory in the absence of further stimulation by antigen.
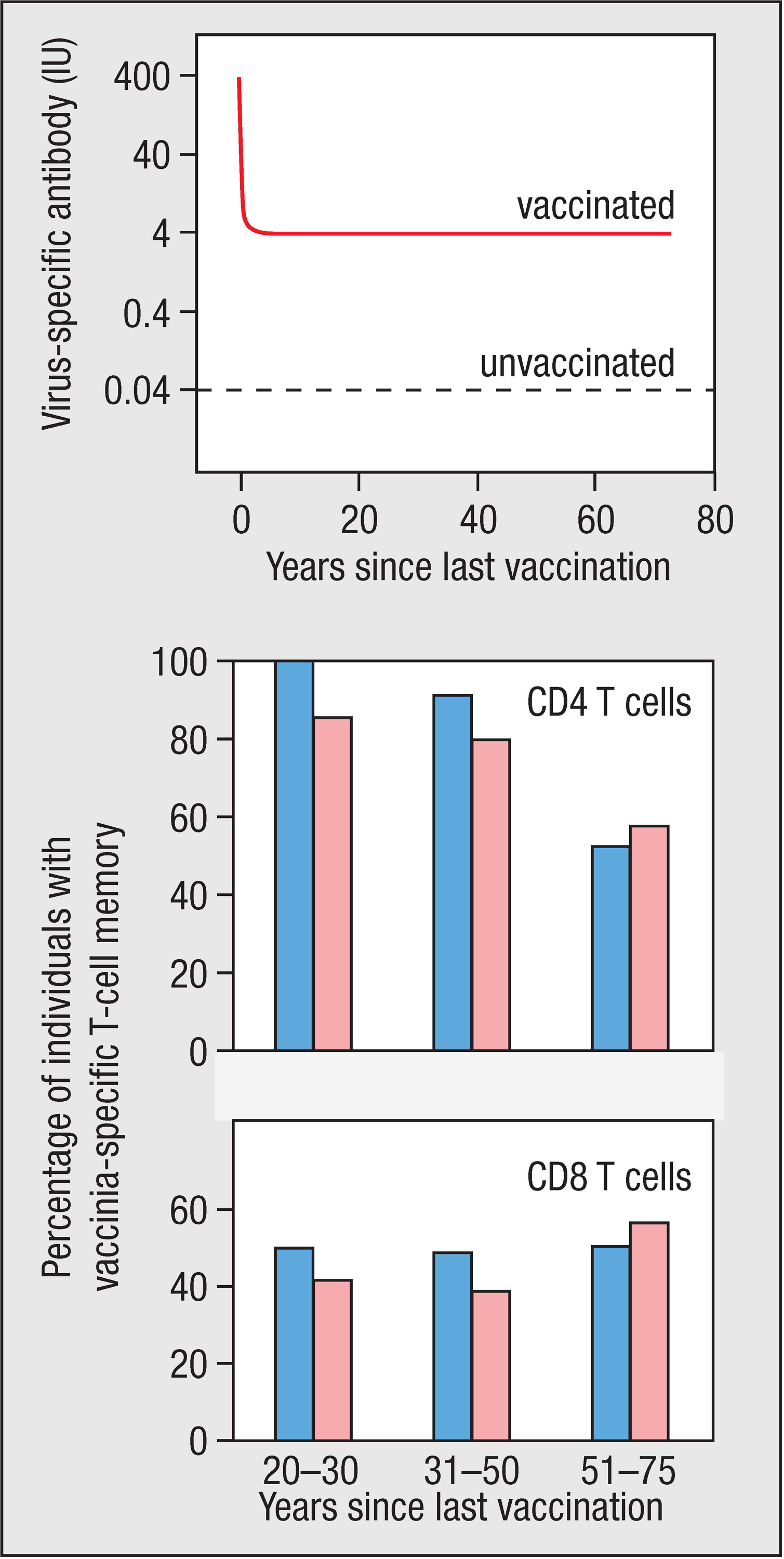
Figure 11.1Retention of vaccinia-specific antibodies and T cells after vaccination against smallpox. Top panel: specific anti-vaccinia antibodies continue to be made for as long as 75 years after the last exposure to vaccinia virus, the smallpox surrogate that is used for vaccination. The numbers represent international units (IU) of antibody, a standardized way of measuring an antibody response. Center and bottom panels: many vaccinated individuals retain populations of vaccinia-specific CD4 T cells and CD8 T cells. Only small differences are observed between individuals who received one (blue bars) or two (pink bars) vaccinations. Courtesy of Mark Slifka.
After vaccination, the amount of vaccinia-specific antibody in the blood increases rapidly to a maximum level and then, over the next 12 months, decreases to about 1% of the maximum. This steady-state level is maintained for 75 years or more, and probably for life (Figure 11.1, top panel). Antibodies only survive in the circulation for about 6 weeks: the steady-state level is maintained by long-lived plasma cells that continue to make vaccinia-specific antibody. After vaccination, the number of virus-specific B cells in the blood also increases rapidly to a maximum and then declines over a 10-year period to reach a stable level, about 10% of the maximum. This pool of cells comprises memory B cells and is maintained for life in a state that can respond to further immunization with vaccinia virus. Vaccination also produces populations of memory CD4 T cells (see Figure 11.1, center panel) and memory CD8 T cells (see Figure 11.1, bottom panel) that similarly persist for 75 years or more, and can respond to vaccinia and smallpox antigens.
Not all forms of protective immunity are as persistent as those induced by the smallpox vaccine or measles infection. After vaccination against diphtheria, a bacterial pathogen, the level of protective anti-diphtheria antibodies in the blood continues to decrease and is halved after 19 years. This compares with 200 years, the estimated half-life of anti-measles protection. In 1918, after cessation of the First World War, an unprecedented influenza pandemic killed between 50 million and 100 million people. A 2008 study of 32 individuals who were born before 1916 found that they all had circulating antibodies specific for the strain of influenza that caused the pandemic. For these individuals, their immunological memories of the influenza virus had lasted for more than 90 years. By contrast, individuals who were born after the pandemic had no antibodies specific for the 1918 pandemic strain of influenza virus.
11-2Antibodies made in a primary response persist in the circulation to prevent reinfection
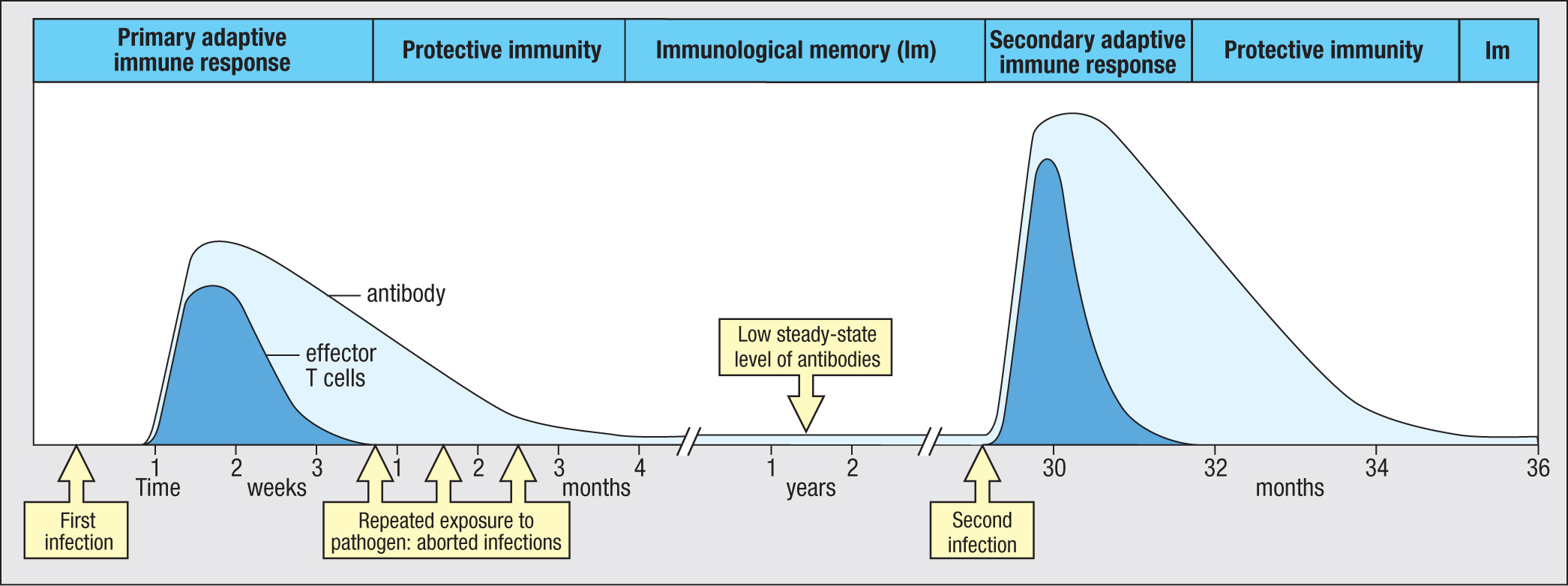
At the termination of an infection by the primary immune response, raised levels of high-affinity pathogen-specific antibodies are present throughout the blood, lymph, and tissues, or at every mucosal surface. The antibodies are secreted by plasma cells residing in the bone marrow or in the tissue beneath a mucosal surface and are sustained at high levels for several months after the infection has been cleared (Figure 11.2). During this time, these antibodies provide protective immunity, ensuring that subsequent contact with the pathogen does not cause disease.
Figure 11.2History of infection by a pathogen. A child’s first infection with the pathogen was not stopped by innate immunity, so a primary immune response was made. Production of pathogen-specific effector T cells and antibodies terminated the infection. The effector T-cell response (dark blue) was soon over, but antibody persisted (light blue), providing immunity that prevented reinfection despite frequent exposure to infected classmates in kindergarten. After 1 year, antibody levels had dropped to a low steady-state level that still provided protection by opsonizing the pathogen for phagocytosis. In the third year after the first infection, the pathogen invaded and established a second infection. The secondary immune response to the pathogen was faster and stronger than the primary immune response, being mediated by high-affinity pathogen-specific IgG, memory T cells, and memory B cells. Because the child’s immune system had retained a ‘memory’ of the first infection, the secondary immune response eliminated the pathogen before there was significant disruption of tissue or symptoms of disease.
Many infectious diseases are seasonal. So during the winter you can be exposed repeatedly to the same cold virus over a period of weeks or months as it passes among family, friends, colleagues, and the community at large. During this time, antibodies raised against a cold caught early in the season prevent any later reinfection with the same virus. On reinvasion, the virus becomes immediately coated with virus-specific IgA or IgG. Neutralized by its coating of antibody, the virus fails to infect cells and replicate (see Section 9-15). In these circumstances, where specific antibody cooperates with the effector functions of innate immunity, a pathogen gets little opportunity to grow and replicate. Consequently, the pathogen load does not reach a point where a secondary adaptive immune response is needed (see Figure 11.2).
Most plasma cells made in the primary response are short-lived effector cells. Their purpose is to proliferate as quickly as possible and to secrete as much antibody as possible. The stress and speed of this activity causes cellular damage and genomic mutation, characteristics that are undesirable in a memory B cell. For this reason these plasma cells are short lived. They die when immune complexes of specific antibody and pathogen antigens bind to FcγRIIB1 on the plasma-cell surface and induce apoptosis of the plasma cells. Consequently, the level of circulating pathogen-specific antibody gradually decreases over a period of 1 year but then reaches a low, steady-state level (see Figure 11.2).
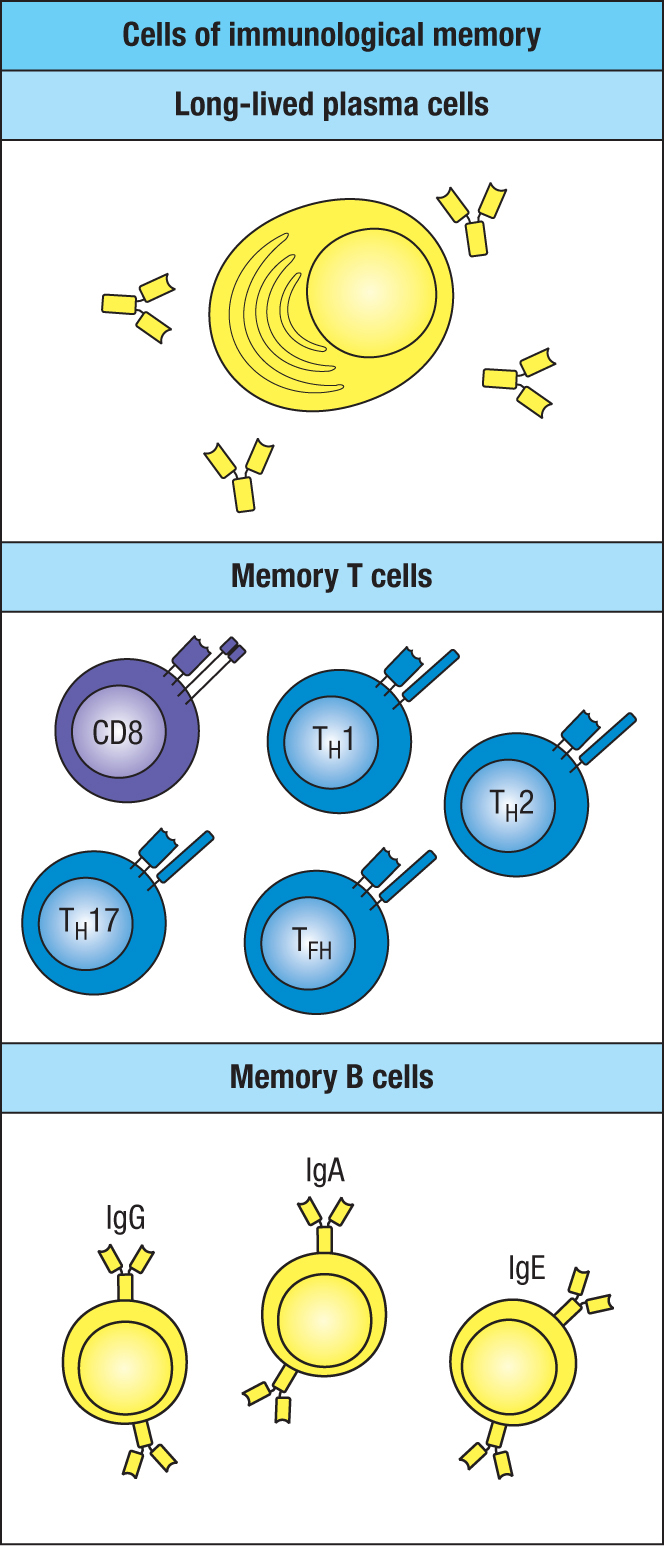
Figure 11.3Cells of immunological memory.
During the later stages of a primary immune response, when the pathogen is defeated and no longer a threat, small clones of long-lived plasma cells are produced under conditions that do not induce cellular damage or genomic mutation. Whereas the rapid proliferation of short-lived plasma cells can introduce undesirable mutations in their rearranged immunoglobulin genes, that problem is avoided during the limited and highly regulated proliferation of the long-lived plasma cells. These plasma cells, which make the most-effective antibodies, are sheltered and nurtured in the bone marrow. The population of long-lived plasma cells is the source of the antibodies that make up the circulation’s steady-state level of antibody. Survival of the long-lived plasma cells does not depend on antigen but is sustained by interactions with bone-marrow stromal cells and by the IL-6 the stromal cells secrete.
Long-lived plasma cells and the antibodies they make constitute one arm of a person’s immunological memory of pathogens; the other arm comprises the memory B cells and T cells (Figure 11.3). After a primary immune response has been made, any subsequent invasion by the same pathogen is confronted by specific, high-affinity antibody from the moment the pathogen enters the body. On binding to the pathogen, the antibody speeds its delivery to phagocytes or other effector cells of innate immunity. In almost all circumstances, these mechanisms are sufficient to terminate the infection. But, when that is not the case, presentation of the pathogen’s antigens by professional antigen-presenting cells will initiate the secondary adaptive immune response.
11-3Memory B cells, naive B cells, and plasma cells are distinguished by the expression of their B-cell receptors
Primary B-cell responses are made by naive B cells, which have surface IgM as their antigen receptor. In the course of the primary response, antigen-activated B cells change the isotype of their antigen receptor by switching the heavy chain from μ to α, γ, or ε. In addition, the affinity of the antibody is increased through somatic hypermutation of the heavy- and light-chain V regions, followed by antigen-mediated selection (see Section 9-7). Because of these changes, none of the B cells that emerge from the primary immune response uses IgM as its B-cell receptor. These B cells, which have experience of a pathogen, are called memory B cells. They use IgG, IgA, or IgE as their B-cell receptor. Plasma cells, the effector cells of the B-cell lineage, are distinguished from naive and memory B cells by not expressing a cell-surface form of immunoglobulin.
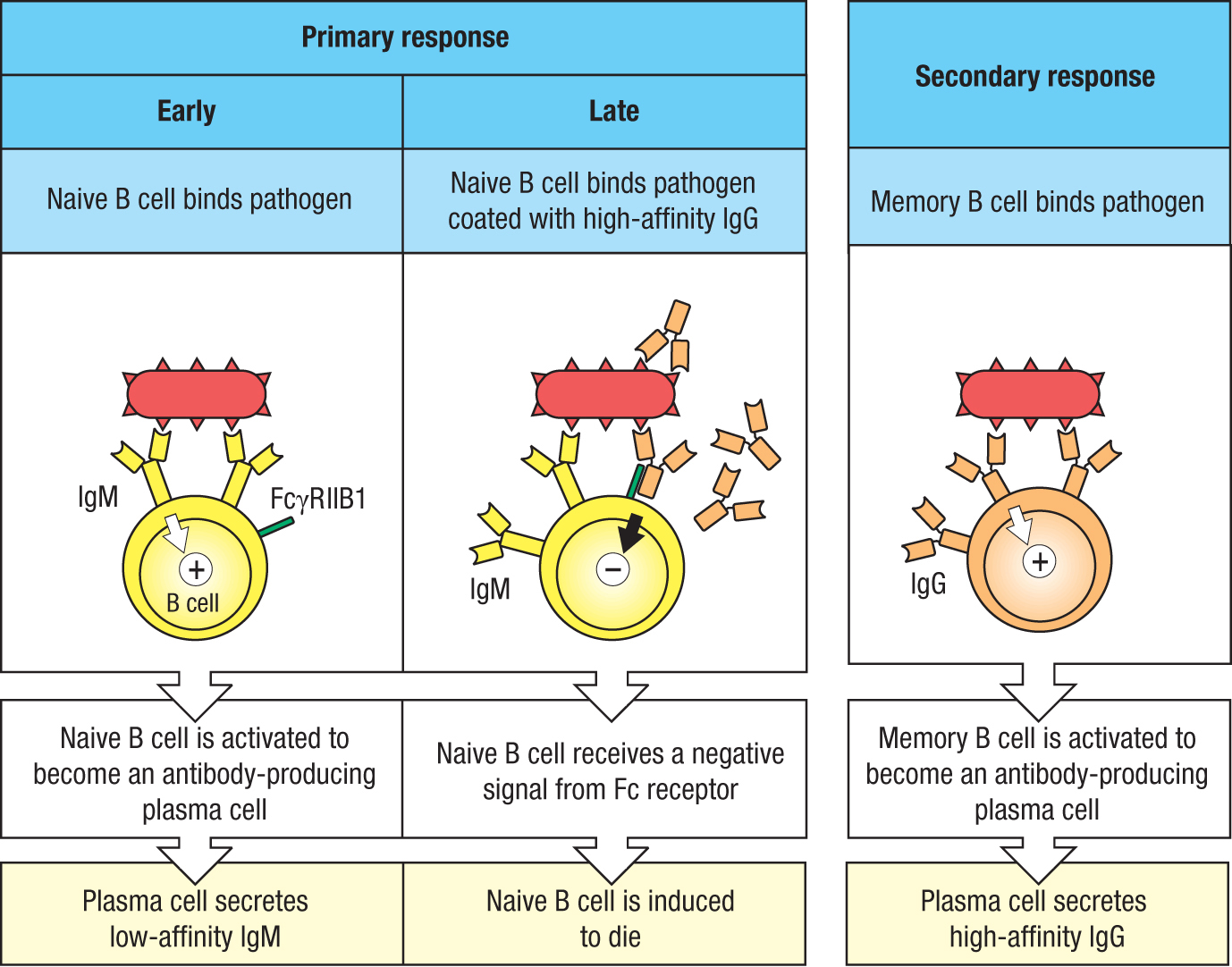
At the beginning of a primary response, only low-affinity IgM antibodies are made, but with the onset of isotype switching, somatic hypermutation, and affinity maturation, IgM gives way to high-affinity IgG. Contributing to this transition is an active mechanism of suppression that prevents any further activation of naive B cells. It involves a pathogen-specific naive B cell simultaneously making two different interactions with an IgG-coated pathogen. The first interaction is between the IgM of the B-cell receptor and its specific antigen on the pathogen surface; the second interaction is between the naive B cell’s inhibitory FcγRIIB1 receptor and the Fc region of IgG bound to its antigen on the pathogen’s surface. Engagement of the FcγRIIB1 by the immune complex of IgG bound to the pathogen delivers an inhibitory signal to the naive B cell that prevents its activation and condemns it to die by apoptosis. The effect of this suppressive mechanism is to cause an overall increase in the affinities of the pathogen-specific antibodies. Memory B cells have high-affinity B-cell receptors and they do not express FcγRIIB1 (Figure 11.4).
Figure 11.4Complexes of IgG and polymeric antigen prevent the activation of naive B cells by cross-linking the B-cell receptor to the inhibitory FcγRIIB1 receptor. In the early stage of a primary immune response, a pathogen binding to the antigen receptor of a naive B cell delivers a signal that activates the cell to become an antibody-producing plasma cell (left panel). At a later stage, the B-cell receptor and the inhibitory Fc receptor (FcγRIIB1) on a naive B cell can be cross-linked by a pathogen coated with IgG, delivering a negative signal that prevents the naive B cell from being activated (center panel). Memory B cells activated in a secondary response do not express FcγRIIB1 and are activated by the pathogen binding to the B-cell’s antigen receptor, cell-surface IgG. Most memory B cells make IgG1 (right panel).
11-4Immune complex–mediated inhibition of naive B cells is used to prevent hemolytic anemia of the newborn
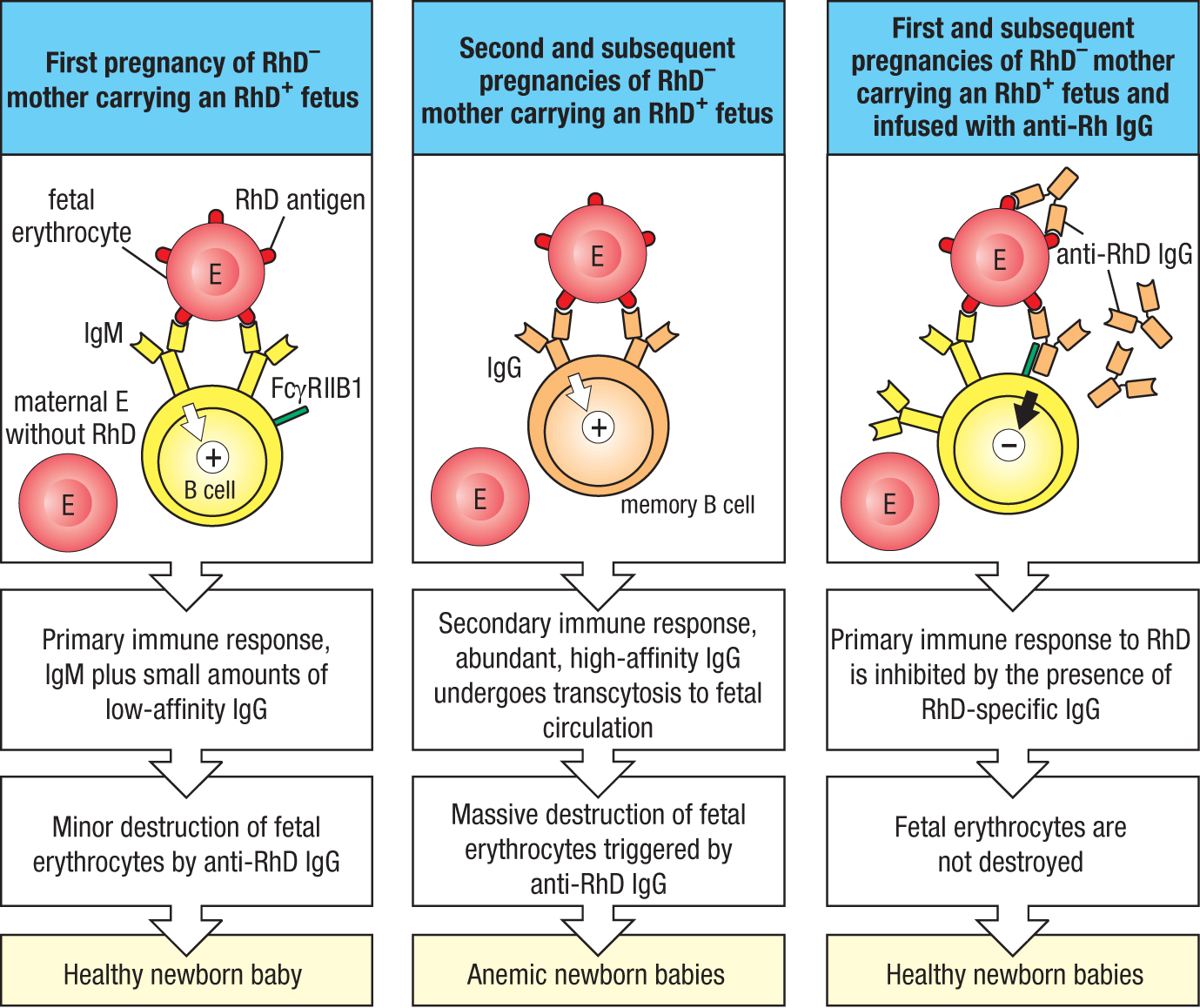
The inhibition of naive B cells by immune complexes and FcγRIIB1 described above (see Section 11-3) is put to practical use in preventing hemolytic anemia of the newborn, also called hemolytic disease of the newborn. This syndrome affects families in which the father has the Rhesus D (RhD) antigen on his erythrocytes and is RhD+, whereas the mother lacks the antigen and is RhD–. During her first pregnancy carrying an RhD+ fetus, fetal erythrocytes cross the placenta and stimulate the mother’s immune system to make anti-RhD antibodies. The antibodies made during this first exposure to RhD cause little harm to the fetus, because they are mostly low-affinity IgM that cannot cross the placenta from mother to child (Figure 11.5, left panel). During a second pregnancy with an RhD+ baby, fetal red cells again cross the placenta and induce the mother to make a secondary immune response to the RhD antigen. This produces high-affinity RhD-specific IgG, which is actively carried across the placenta by FcRn (see Section 9-14). The fetal erythrocytes become coated with IgG and thus opsonized are removed from the circulation by splenic macrophages and destroyed. When born, the baby will have severe anemia (Figure 11.5, center panel), which can lead to further complications and can be fatal. Hemolytic anemia of the newborn is prevalent in populations of European origin, in which 16% of mothers are RhD– and 84% of babies are RhD+, but is rare in Africans and Asians, in which fewer than 1% of the population is RhD–.
Figure 11.5Passive immunization with anti-Rhesus D IgG prevents hemolytic anemia of the newborn. In human populations, up to 16% of individuals lack the erythrocyte antigen Rhesus D (RhD). RhD– mothers carrying RhD+ fetuses are exposed to fetal erythrocytes and make RhD-specific antibodies that pass to the fetal circulation and cause fetal erythrocytes to be destroyed. Left panel: in a first pregnancy of this type, the antibodies produced in the primary response are mainly IgM, which does not cross the placenta, plus small amounts of low-affinity IgG that causes only minor damage to fetal erythrocytes, allowing a healthy baby to be born. Center panel: during a second pregnancy, a secondary immune response is made. This produces large amounts of high-affinity IgG, some of which is actively transported from the maternal circulation to the fetal circulation by FcRn. The anti-RhD IgG causes such massive destruction of fetal erythrocytes that the baby is born with anemia. Right panel: this disease is prevented if the mother is passively infused with purified human anti-RhD antibodies during her first and subsequent pregnancies. The immune complexes of fetal erythrocytes coated with IgG prevent the mother from making a primary B-cell response against the RhD antigen.
To prevent hemolytic anemia of the newborn, pregnant RhD– women who have yet to make anti-RhD antibodies are infused, during the 28th week of pregnancy, with a preparation of purified human anti-RhD IgG antibody called RhoGAM. The amount of antibody given is sufficient to coat all the fetal erythrocytes that cross the placenta and enter the maternal circulation. The mother has naive RhD-specific B cells that will recognize the RhD antigen in the complex with IgG. In addition, the Fc of the IgG will bind the B cell’s FcγRIIB1 receptor, delivering inhibitory signals that inactivate the naive B cell. This mechanism causes inactivation of all the mother’s naive RhD-specific B cells (Figure 11.5, right panel).
Tissue injury during the traumatic process of giving birth further exposes the mother to her baby’s red blood cells. To ensure that they do not activate any naive RhD-specific B cells, the mother will receive a second infusion of RhoGAM within 3 days after the birth. Although the amount of infused anti-RhD antibody (300 μg) is far in excess of that needed to coat all fetal erythrocytes in the maternal circulation, almost none of the antibody gets across the placenta and into the fetal circulation. This is because the anti-RhD IgG competes for access to FcRn with the 60 g of circulating maternal IgG that is not specific for RhD.
11-5Long-lived plasma cells are the major mediators of B-cell memory
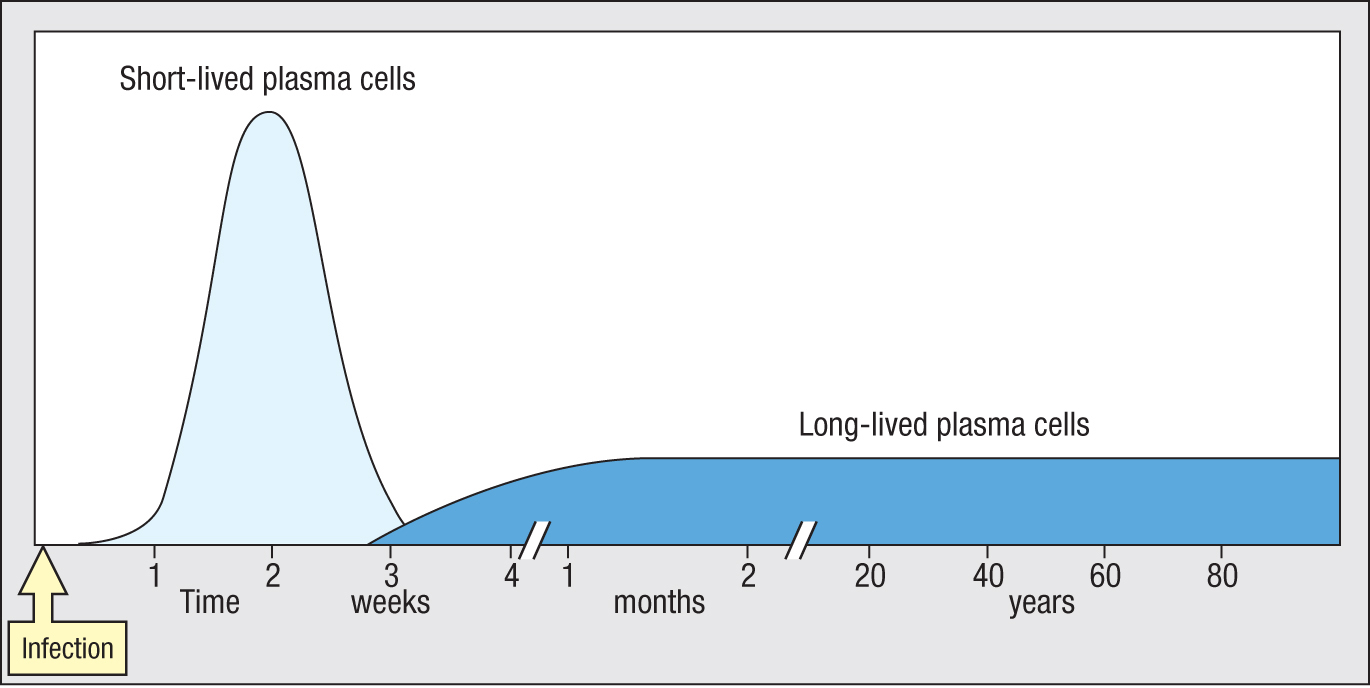
For common and deadly pathogens that cannot evolve rapidly to escape human immunity, such as the measles, mumps, and smallpox viruses, it is advantageous for humans to maintain a lifetime’s supply of antibodies against them. Primary infection by these viruses or vaccination with viral proteins leads to the production of plasma cells secreting neutralizing antibodies against the pathogen. As we saw earlier in the chapter, these plasma cells are of two types. First, short-lived plasma cells are rapidly brought into play to produce a large burst of antibody, which serves to limit the ongoing infection. The long-lived plasma cells derive from B cells that have been refined by isotype switching, hypermutation, and selection by antigen. Antibodies made by the long-lived plasma cells terminate the ongoing infection and are maintained at a low but steady level in the circulation to prevent future infections from taking hold (see Section 11-2). The short-lived plasma cells are present at extrafollicular sites in the secondary lymphoid tissue, whereas the long-lived plasma cells are resident in the bone marrow or the gut. Plasma cells never divide and do not depend on interactions with lymphocytes.
In human bone marrow, four subpopulations of long-lived plasma cells have been distinguished. The subpopulations differ in their relative age. The most useful plasma cells are usually the oldest. For every person studied, all the long-lived plasma cells making antibodies against the slowly evolving mumps and measles DNA viruses have the CD19–CD38hiCD138+ phenotype. That is not the case for plasma cells making antibodies against influenza and other rapidly evolving RNA viruses. By mutating their antigenic sites these viruses escape from human antibodies, which are only effective for a few years. The emerging picture is that the long-lived plasma cells provide a serological memory, an archive of plasma cells representing a person’s history of response to infection and vaccination. The four subpopulations of plasma cells represent cells of increasing age and value. In the mucosal immune system, long-lived plasma cells reside in the gut where they provide defense against enteric pathogens. The B-cell strategy of combating infection first with an army of short-lived plasma cells and second by a refined and selected population of long-lived memory cells (Figure 11.6) is also used by T cells.
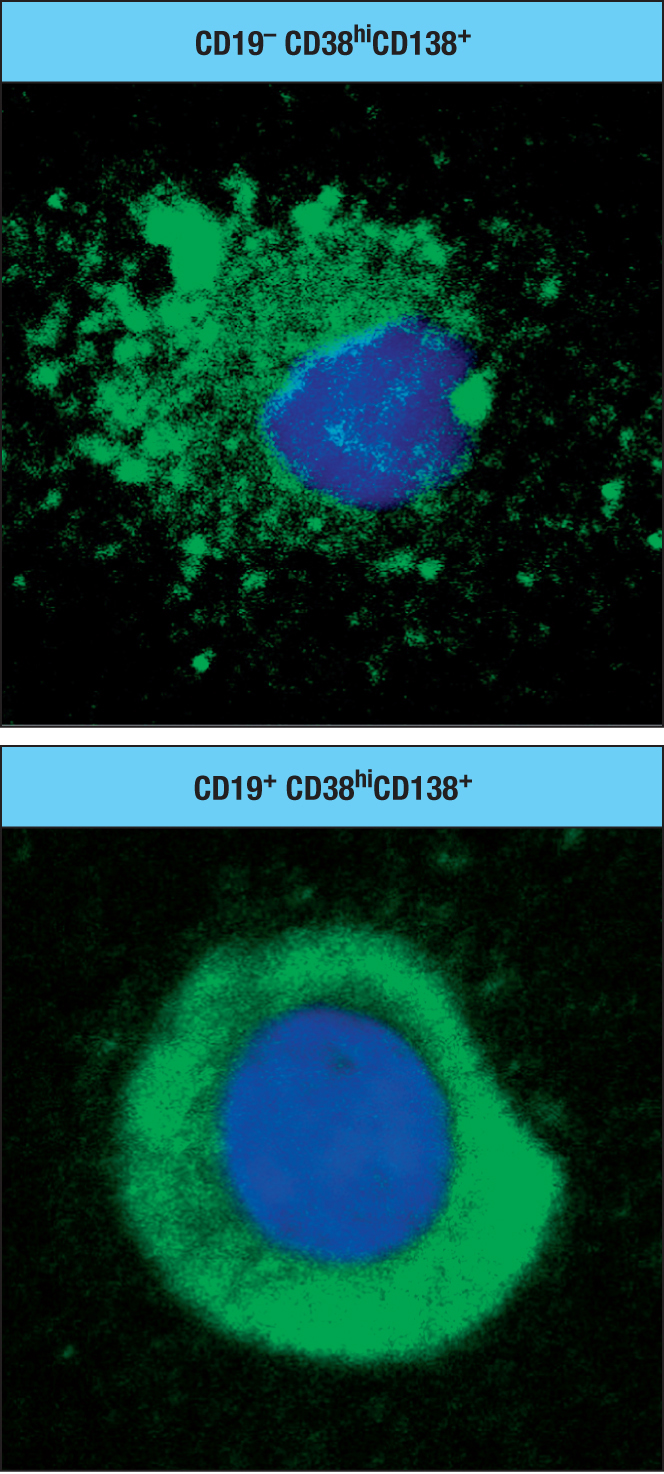
Figure 11.6In responding to infection, a large wave of short-lived plasma cells is followed by a smaller, selected group of long-lived plasma cells.Figure 11.7Active autophagy is a characteristic of long-lived CD19– CD38hiCD138+ plasma cells. Confocal microscopy of a plasma cell of the CD19–CD38hiCD138+subpopulation (upper panel) compared with a plasma cell of the CD19+CD38hiCD138+ subpopulation (lower panel). Note the extensive autophagy in the CD19– plasma cell as seen by the fragmentation of the cytoplasm (green).
Although long-lived plasma cells do not divide, they are metabolically very active. In addition to making and secreting antibodies, they invest heavily in the maintenance of their infrastructure. This is achieved by autophagy—a mechanism by which cellular components are degraded by lysosomal degradation and then renewed. Autophagy is more active among the CD19–CD38hiCD138+ cells than in the other subpopulations of long-lived plasma cells (Figure 11.7).
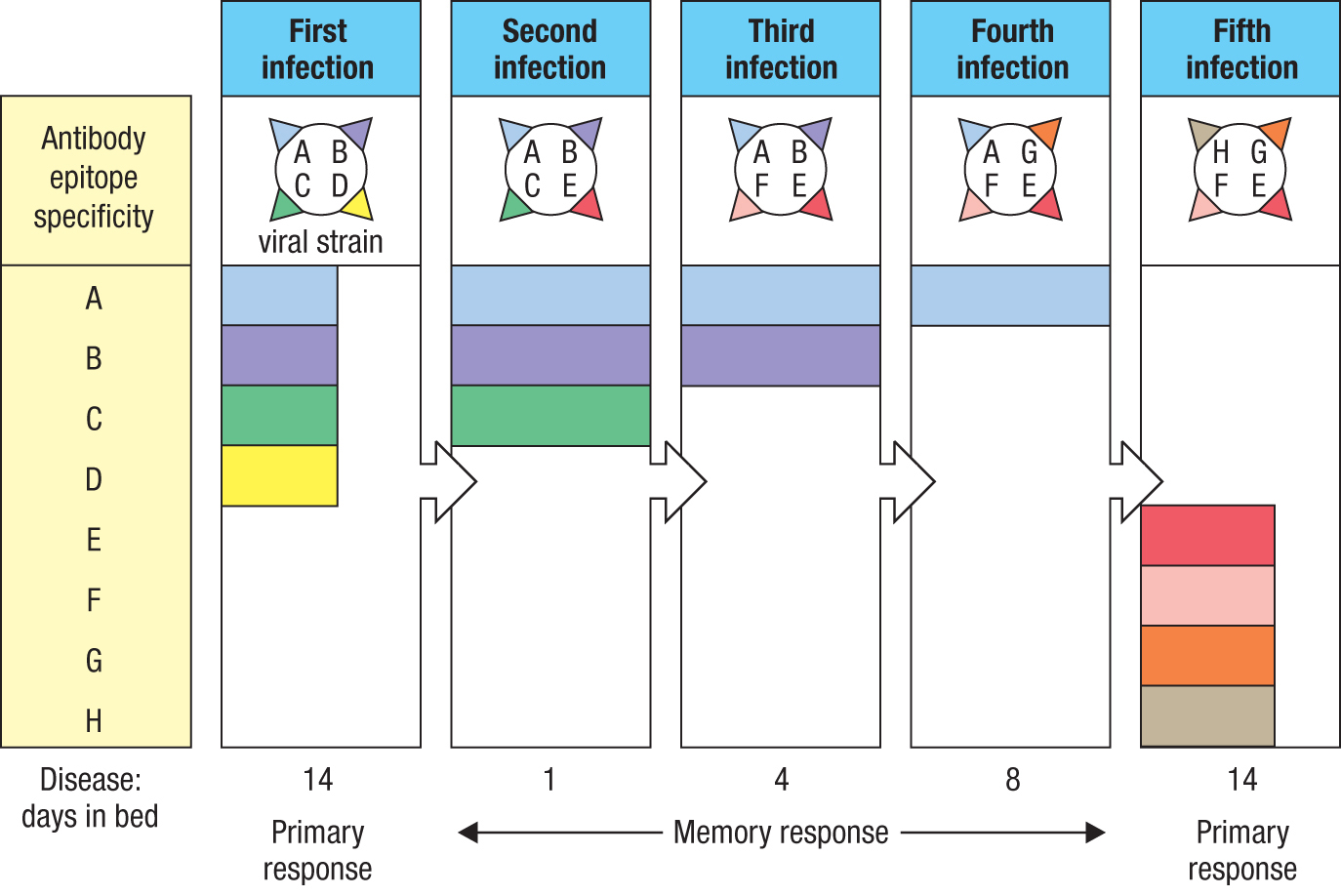
11-6In responses to influenza virus, immunological memory is gradually lost with successive infections
The suppression of naive B-cell activation that occurs during the secondary immune response (see Section 11-3) is an effective strategy against pathogens such as measles virus, which do not vary their antigens. But this approach is disadvantageous when confronting highly mutable pathogens, such as RNA viruses and in particular the influenza virus. Because influenza virus changes its epitopes on a yearly basis, a person’s immunity to influenza virus erodes with time, as the antibodies made during past infections no longer recognize the current viral strains. Because the activation of naive influenza-specific B cells is prevented, new clones of influenza-specific B cells cannot be brought into play until the time is reached when none of the antibodies made in the primary response is able to recognize the current strain of influenza. Thus the imprint made by the first infecting strain is broken only on infection with a strain of influenza that lacks all the B-cell epitopes of the original strain (Figure 11.8). At the population level, this means that every year there are millions of adults who have little or no effective immunity against the latest strains of the virus. Unless they get vaccinated, this loss of immunity inevitably leads to a severe, full-blown case of influenza, which stimulates a primary B-cell response targeted against the full complement of new influenza epitopes.
Figure 11.8Influenza is a highly mutable virus that progressively erodes human immunological memory of influenza. A person’s history of infection with influenza is shown here. The first infection is with a strain of influenza virus that elicits a primary antibody response to viral epitopes A, B, C, and D. The following year the person is infected with a virus that has lost epitope D and gained epitope E. In the second year the person is infected with a virus that has lost epitope C and gained epitope F. In the third year the person is infected with a virus that has lost epitope B and gained epitope G. In the fourth year the person is infected with a virus that has lost epitope A and gained epitope H. Against this virus the person has no memory or immunity and will be infected, become a patient, and suffer the disease of influenza. During infection the patient will make a primary immune response that will provide antibodies and memory that are specific for epitopes E, F, G, and H.
11-7Antigen-mediated activation of naive T cells gives rise to effector and memory T cells
We have seen how activation of naive B cells during the primary response to a pathogen leads to the development of clones of memory B cells that are quicker and more effective than naive B cells at preventing disease when the pathogen next invades. In a similar manner, the activation of naive T cells in the primary response leads to clones of memory T cells with improved capacity for attacking the pathogen. The population of memory T cells mirrors the population of pathogen-specific effector cells that contributed to the primary response made against the pathogen; it can therefore include TFH, TH1, TH2, and TH17 CD4 T cells and CD8 T cells. This congruence between the effector and memory T cells arises because they have a common origin. We will use CD8 T cells as our example here, because they have been studied to a greater extent than the CD4 T cells, but it is clear that the same general principles apply to CD4 T cells.
Naive T cells are long-lived, quiescent cells that survive by engaging in catabolic metabolism, also called starvation metabolism. This involves mitochondrial oxidative phosphorylation, fatty acid oxidation, autophagy (turnover of cellular constituents and organelles), and mitophagy (turnover of mitochondria). Glycolysis is dormant in naive T cells. When the antigen receptor of a naive CD4 or CD8 T cell engages the complex of MHC and peptide antigen presented by a dendritic cell, the T cell begins its activation and is then no longer naive. In preparing to divide, the previously quiescent T cell changes its metabolism from catabolism to anabolism. Obtaining energy from glycolysis enables the T cell to synthesize and assemble the macromolecular complexes that are necessary for mitotic cell division. This change, described as the metabolic reprogramming of the T cell, is orchestrated by mTORC1 (mammalian target of rapamycin complex 1), a protein kinase complex in the cytoplasm. mTORC1 activates the lysosomes to increase their degradation of macromolecules and the uptake by the cell of the many small-molecule nutrients, such as amino acids, that are needed to synthesize proteins, nucleic acids, and other T-cell macromolecules. In addition, autophagy and mitophagy are inhibited.
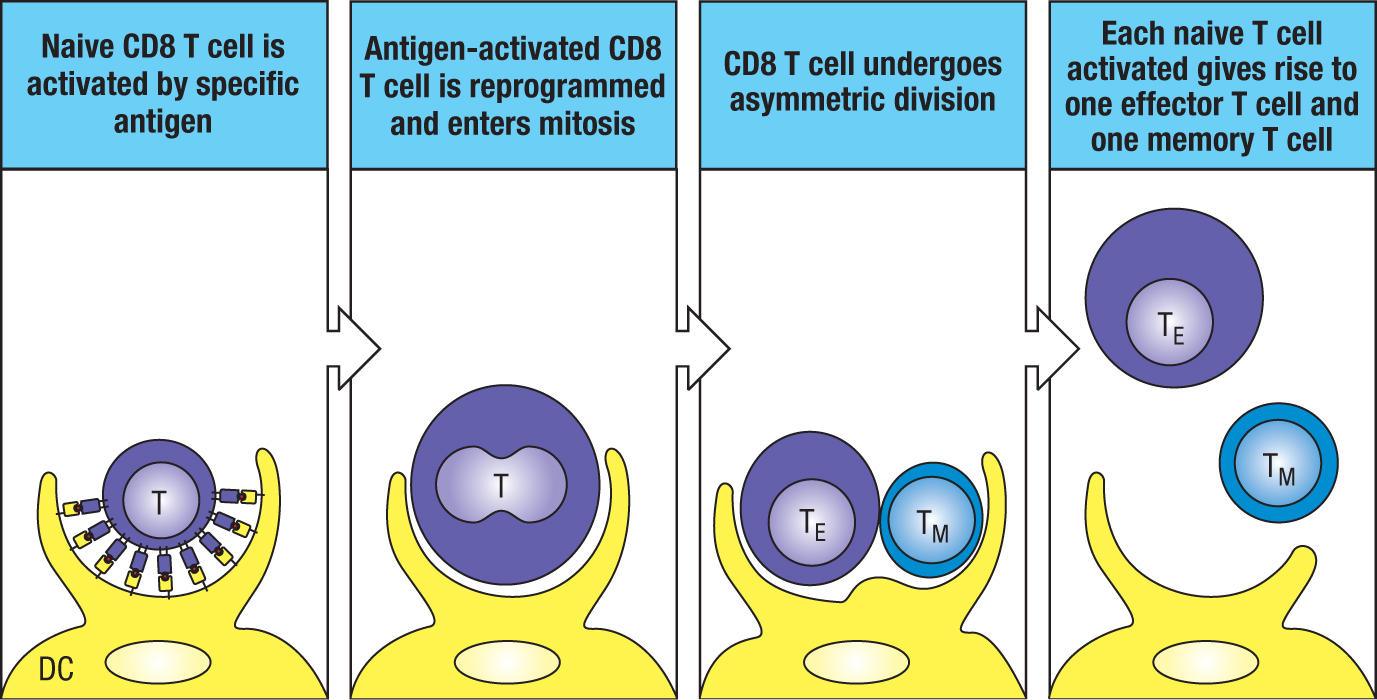
Mitosis occurs while the antigen-activated T cell remains attached to the antigen-presenting dendritic cell. After the first cell division, one of the two daughter cells becomes physically closer to the dendritic cell than the other (Figure 11.9). In the course of mitosis, most cellular components and contents are divided equally between the two daughter cells, but mTORC1 is preferentially inherited by the daughter cell closest to the dendritic cell (Figure 11.10). The consequence of this preference is that the proximal daughter cell inherits the active metabolic program of the mother and undergoes further cell divisions to form a clone of effector CD8 T cells. By contrast, the distal daughter cell has a metabolism closer to that of a naive T cell and is likely the precursor of a memory cell. Correlating with these metabolic differences, the proximal daughter inherits more CD8 from the mother than the distal daughter does. The process by which mitosis of a mother cell produces nonidentical daughters is called asymmetric division.
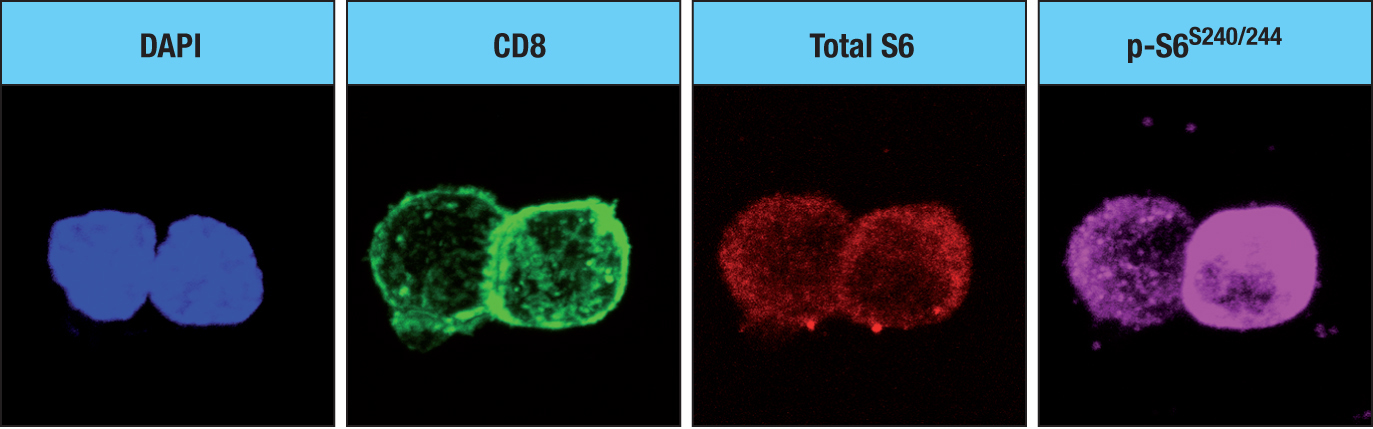
Figure 11.9Antigen activation of a naive CD8 T cell. On activation, the naive CD8 T cell becomes metabolically reprogrammed. It then undergoes asymmetric division to give one daughter cell that is an effector T cell (TE) and one that is a memory T cell (TM). DC, dendritic cell.Figure 11.10When a naive T cell is activated by antigen, it undergoes an asymmetric division. The fluorescence micrographs show an activated naive T cell that is undergoing its first, and asymmetric, division. Four fluorescent stains show the distribution of different cellular components. The chemical DAPI (4′,6-diamidino-2-phenylindole) stains regions of genomic DNA that are rich in adenine and guanine (blue). CD8 is stained with anti-CD8 antibody and thus shows the co-receptor CD8 (green). Total S6 shows the distribution of ribosomal protein S6 (red), which is similar in the two daughter cells; p-S6S240/244 (pink) shows the distribution of the S6 protein that has been phosphorylated on serine residues 240 and 244, which correlates with the activity of mTORC1. The proximal cell (see text) is on the right. Micrographs courtesy of Jonathan Powell.
At the beginning of the primary CD8 T-cell response to a viral infection, the immediate urgency is to produce a large clone of cytotoxic T cells. Although the first few divisions of an activated T cell can be asymmetric, as the response gains momentum the divisions become symmetric and faster, giving rise to an army of thousands of cytotoxic T cells. As we saw in B-cell activation, the effector CD8 T cells in this first burst of the immune response are subjected to stress and mutation and have short lives. Once the pathogen is being defeated, clones of slow-dividing and carefully maintained CD8 T cells emerge to form a population of long-lived memory CD8 T cells that is analogous to the population of memory B cells. This involves a reprogramming from anabolic to catabolic metabolism that permits these long-lived cells to self renew and thus remain healthy.
Like effector CD8 T cells, the TH1, TH2, and TH17 CD4 cells travel to the infected tissue, where they proliferate to produce expanded clones of effector cells (see Chapter 8). By contrast, the TFH CD4 cells proliferate in the secondary lymphoid tissue where they provide help to antigen-specific B cells (see Chapter 9).
11-8Two subpopulations of circulating memory cells patrol different tissues of the body
The two subsets of circulating memory T cells have been distinguished by the tissues in which they are found and respond to antigen. Central memory T cells (TCM cells) express L-selectin (CD62L) and the chemokine receptor CCR7, which allows them, like naive T cells, to enter secondary lymphoid organs and survey the antigens presented there by dendritic cells. Central memory T cells have limited capacity for effector function, but they combine this with a low threshold for activation and a high capacity to produce IL-2, proliferate, and differentiate into effector T cells. During the secondary immune response, memory B cells in secondary lymphoid tissue process and present the pathogen’s antigens to memory TFH cells in the same way as in the primary response. This leads to germinal center reactions in which another round of somatic hypermutation and selection by antigen further increases the affinity of the pathogen-specific antibodies.
Lacking L-selectin and CCR7, effector memory T cells (TEM cells) are excluded from secondary lymphoid tissues. Instead, they express other chemokine receptors, such as CCR6, CCR4, CXCR3, and CCR5, by which they gain entry to non-lymphoid tissues, notably inflamed tissues and mucosal tissues. Effector memory T cells are heterogeneous and represent the CD8 and CD4 TH1, TH2, and TH17 subsets of primary effector cells. In patrolling the peripheral tissues, effector memory T cells can respond immediately to an infection at its site of origin. This capability is complemented by the slower activation of central memory T cells in draining lymphoid tissue, which eventually produces more of the effector T cells.
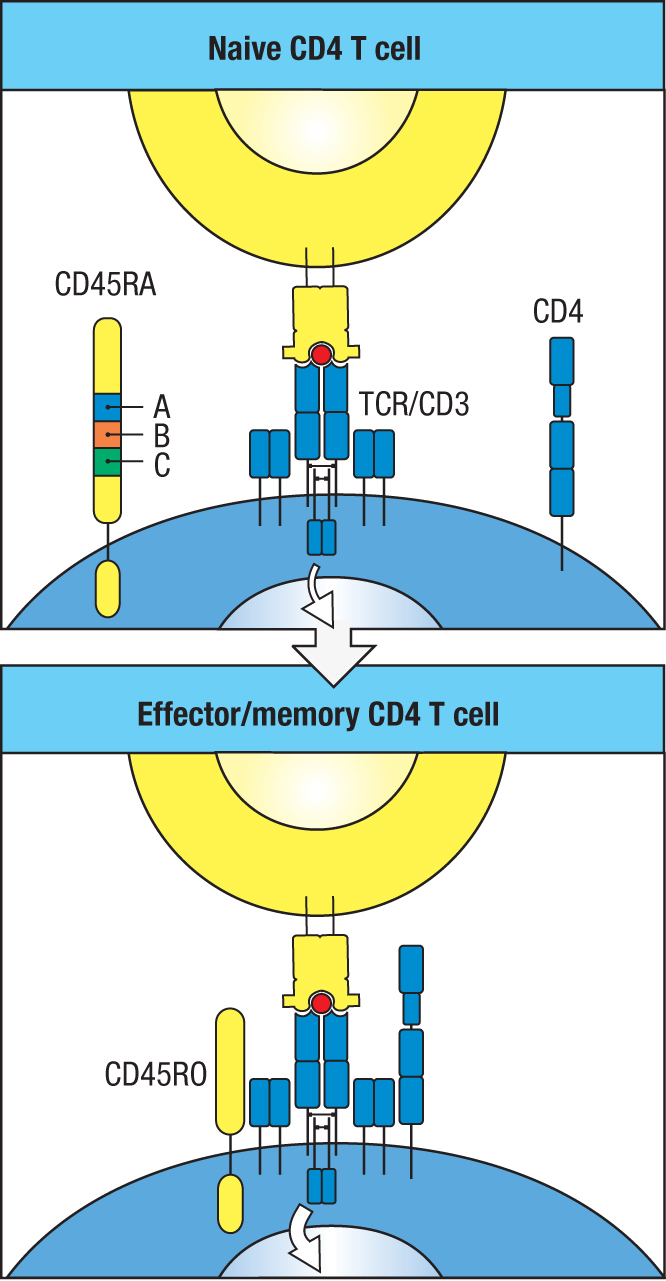
Naive, effector, and memory T cells exhibit similar expression patterns for most of their expressed genes. Apart from CD4 and CD8 and the effector molecules that distinguish CD4 and CD8 T cells, the main differences are in the proteins involved in cell activation, migration, and signaling, and also in cytokines, chemokines, and their receptors (Figure 11.11). A major consequence of these differences is that effector cells and memory cells are more readily activated than naive cells. Contributing to this difference is CD45, a tyrosine phosphatase involved in signaling from the T-cell receptor. Because of alternative mRNA splicing, the extracellular domain of CD45RO is 381 amino acids shorter than that of CD45RA, which allows better interactions with the T-cell receptor. The expression of CD45RA by naive T cells therefore contributes to the higher threshold for their activation compared to that for the effector and memory T cells that express CD45RO (Figure 11.12). Another key feature that distinguishes the activation of memory T cells from that of naive T cells is that it does not depend on CD28-mediated co-stimulation. This property, which is shared with effector T cells (see Figure 8.20, p. 233), has the effect of accelerating the activation of memory T cells.
Figure 11.11Naive, effector, and memory T cells differ in cell-surface phenotype.
Protein
Function
Naive
Effector
Memory
CD44
cell adhesion
+
+++
+++
CD45RO
tyrosine phosphatase
+
+++
+++
CD45RA
tyrosine phosphatase
+++
+
-
CD69
activation marker
-
+++
-
IFN-γ
cytokine
-
+++
+++
CD25
IL-2 receptor
-
++
-
CD127
IL-7 receptor
++
-
+++
Ly6C
surface marker
+
+++
+++
CXCR4
chemokine receptor
+
++
++
Figure 11.12Different isoforms of CD45 make memory T cells easier to activate than naive T cells. CD45 is a transmembrane tyrosine phosphatase involved in T-cell activation. Naive T cells express the CD45RA isoform (upper panel), whereas memory T cells express the CD45RO isoform (lower panel). As a consequence of differential mRNA splicing, CD45RA is bigger than CD45RO and less able to interact with the T-cell receptor complex and activate T cells. The additional 381 amino acid residues present in CD45RA are encoded by exons A, B, and C (upper panel).
11-9Primary infections of a non-lymphoid tissue produce resident memory T cells that live within the tissue
All primary immune responses are initiated in secondary lymphoid tissue. For example, in a skin infection, dendritic cells carry the pathogen’s antigens to the draining lymph node (see Figure 8.1, p. 214), where they are presented to antigen-specific naive T cells. Upon antigen-mediated activation, each naive T cell gives rise to a large clone of effector T cells that migrate to the infected skin to clear the infection. These short-lived effector cells cooperate to clear the infection and then die by apoptosis in the tissue. As the infection and the primary immune response subside, small clones of slowly proliferating memory cells (see Section 11-7) enter the damaged tissue, which is now being repaired. As part of that repair, the pathogen-specific memory T cells are incorporated as sentries placed throughout the repaired tissue. These cells are absent from tissue that was not infected, and they stay in the skin long after infection has gone and the tissue has healed. These long-lived cells, called resident memory T cells (TRM cells), never return to the circulation but stay to protect the skin, and that is where they die.
The benefit of populating non-lymphoid tissues with resident memory T cells during a primary immune response is that it reduces the time taken to initiate a secondary immune response against subsequent skin infection by the same pathogen. Unlike the primary response, when antigen and naive lymphocytes must travel to the draining lymph node and effector cells must travel from the node to the skin, the presence of resident memory T cells in the skin means that all the necessary interactions can occur at or near the site of infection. Onset of a second infection in the skin leads to an inflammatory innate immune response that activates a secondary immune response from the resident memory T cells. Further improving the defense, after experiencing one skin infection by a pathogen, a subsequent infection with the same pathogen at another area of skin will mobilize the existing resident memory T cells to travel to the site of the current infection and act there to eliminate the pathogen (Figure 11.13).
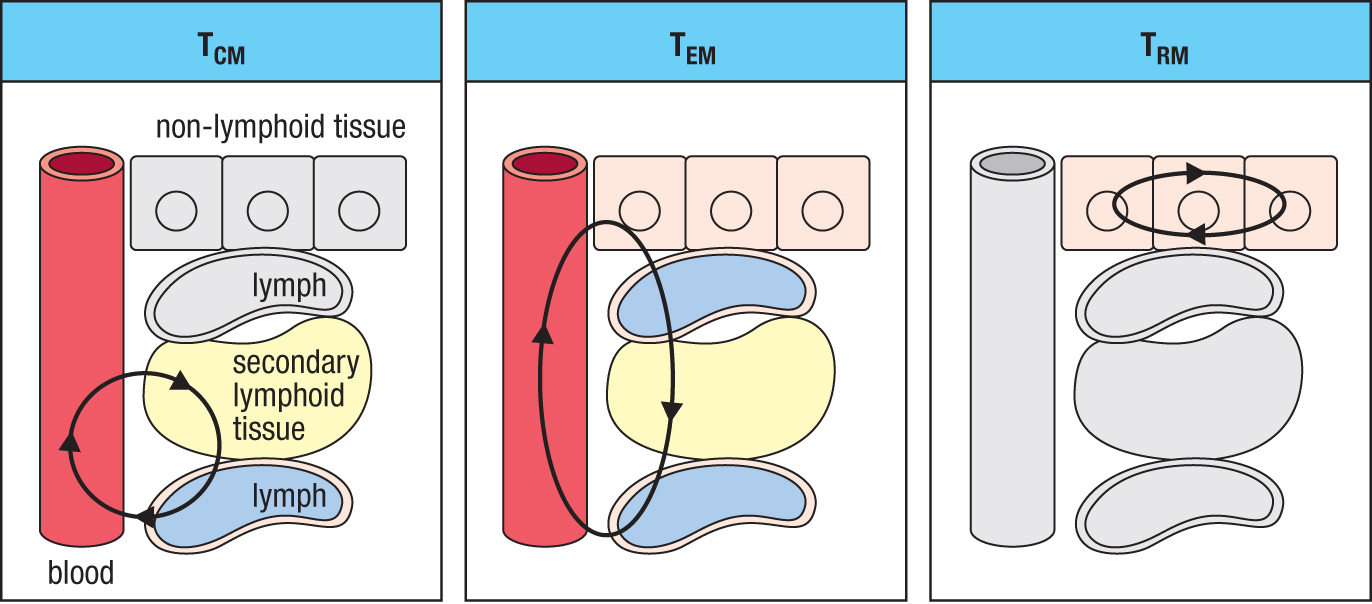
Figure 11.13Three types of memory T cell have different patterns of migration through tissues. For each type of cell, the tissues it circulates through are in color; the other tissues are gray. Like naive T cells, central memory T cells (TCM cells) circulate between the blood, lymph, and secondary lymphoid tissues. Effector memory T cells (TEM cells) circulate from blood to non-lymphoid tissues and then return in the lymph to the blood. Resident memory T cells (TRM cells) are based in a non-lymphoid tissue, where they can rapidly respond to local infections.
11-10Resident memory T cells are the most numerous type of memory T cell
The skin of a healthy human is estimated to contain about 20 billion memory T cells, which is 50-fold the number of circulating T cells in blood and lymph. All resident memory T cells in the skin express the CD45RO marker of memory cells (see Figure 11.12) as well as CLA and CCR4, the skin-homing addressins. They also have diverse T-cell receptor repertoires, consistent with the memory cells having been selected by a wide range of invading microorganisms. TRM cells are also present in similarly large numbers in the gut, the lungs, and the reproductive tract. These tissues are collectively known as epithelial barrier tissues, because they define and separate ecologically different microbial environments in the human body.
Summary
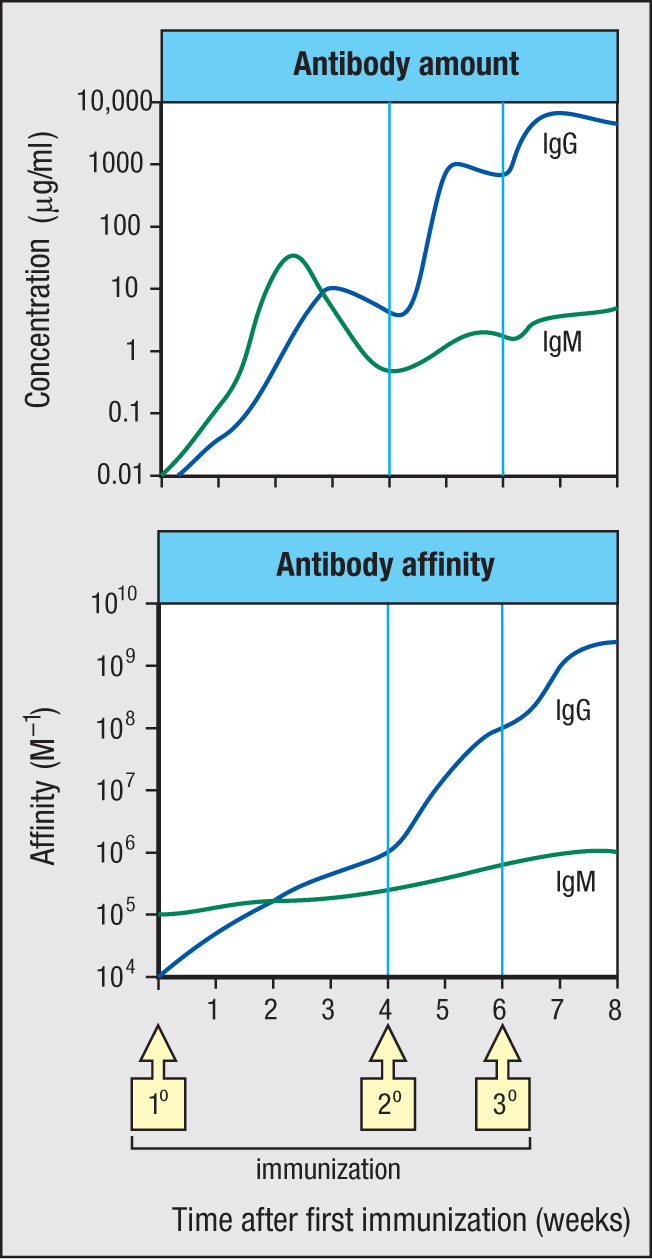
The primary immune response to a pathogen comprises a first phase of innate immunity and a second phase of adaptive immunity. When successful, the primary immune response serves three purposes: it clears the infection, temporarily strengthens defenses to prevent reinfection, and establishes a long-lasting immunological memory of the pathogen. This memory ensures that subsequent infections with the same pathogen will provoke a secondary immune response that is faster, stronger, and more effective than the primary response. In the secondary response, new antibody is detectable in the blood after only 4 days, compared with 8 days in the primary response. In the example of an immune response shown in Figure 11.14, the quantity and affinity of IgG made in the secondary response is orders of magnitude higher than that of IgG made in the primary response. Numerous factors synergize with each other to achieve these improvements (Figure 11.15). Immunological memory is mediated by long-lived populations of memory plasma cells that continue throughout life to make small amounts of high-affinity pathogen-specific antibodies and by long-lived populations of pathogen-specific memory B cells and memory T cells. In the secondary response, the lymphocytes and effector mechanisms of adaptive immunity are deployed from the very start of infection and in greater force than was possible in the primary response. Throughout the secondary response, innate immunity works in partnership with adaptive immunity, which decreases both the time taken to clear the infection and the collateral damage to tissues caused by inflammation. Because of the efficiency and effectiveness of the secondary immune response, the disease caused by a second infection with a pathogen is shorter and less severe than that arising from a first infection and is often undetectable. In the course of the secondary response, further improvements to pathogen-specific immunity and memory are made, which decrease the probability of future infection by that pathogen.
Figure 11.14The amount and affinity of antibody increase after successive immunizations with the same antigen. This figure shows the results of an experiment using mice that mimics the development of specific antibodies when a person is given a course of three immunizations (1°, 2°, and 3°) with the same vaccine. The upper panel shows how the amounts of IgM (green) and IgG (blue) present in blood serum change over time. The lower panel shows the changes in average antibody affinity that occur. Note that the vertical axis of each graph has a logarithmic scale because the observed changes in antibody concentration and affinity are so large.
Figure 11.15Differences between the primary and secondary immune responses.
Differences between primary and secondary immune response to a pathogen
Primary response
Secondary response
A small number of pathogen-specific cells
A large number of pathogen-specific cells
Delay before specific antibodies are made
Specific antibodies are already present
Starts with IgM of low to medium affinity
Antibodies are isotype-switched and of high affinity
High threshold of activation
Low threshold of activation
Delay before effector T cells are activated and enter infected tissues
Effector T cells are present and activated in the infected tissue
Innate immunity works alone until an adaptive response is activated and ongoing
There is close cooperation between innate and adaptive immunity from the start of infection
the specific immunological resistance to a pathogen that is present in an individual during months after either vaccination or recovery from an infection with the pathogen, and which is due to pathogen-specific antibodies and effector T cells produced during the primary response. (Chapter 1, Chapter 11)
type of long-lived CD4 T cell that is produced from activated T cells during the primary response to an antigen. On subsequent exposure to their specific antigen, memory CD4 T cells are activated to differentiate into effector CD4 T cells as part of the secondary and subsequent immune responses to that antigen. (Chapter 11)
type of long-lived CD8 T cell that is produced from activated T cells during the primary response to an antigen. On subsequent exposure to their specific antigen, memory CD8 T cells are activated to differentiate into cytotoxic CD8 T cells as part of the secondary and subsequent immune responses to that antigen. (Chapter 11)
type of plasma cell produced toward the end of an adaptive immune response that can last a lifetime, producing a steady level of protective antibody. They reside in the bone marrow. (Chapter 11)
a potentially fatal disease caused by maternal IgG antibodies directed toward paternal antigens expressed on fetal red blood cells. The usual target of this response is the RhD blood group antigen. Maternal anti-RhD IgG antibodies cross the placenta to attack the fetal red blood cells. Also known as erythroblastosis fetalis. (Chapter 11)
blood group antigen belonging to the Rhesus group. It must be matched for successful blood transfusion. RhD mismatch between fetus and mother is the cause of hemolytic anemia of the newborn. (Chapter 11)
immunological memory maintained by long-lived plasma cells, an archive of plasma cells representing a person’s history of response to infection and vaccination. (Chapter 11)
the degradation of a cell’s own components in its lysosomes, enabling cellular building blocks to be reused. It is a mechanism of cell maintenance. (Chapter 11)
a cell division that produces two daughter cells that are unlike each other in some way; for example, one may be smaller than the other or one may contain a different complement of cytoplasmic proteins than the other. (Chapter 11)
one of two subsets of circulating memory T cells (the other being effector memory T cells) that are distinguished by different activation requirements. Central memory T cells have a preference for the T-cell zones of secondary lymphoid tissues and take longer than effector memory T cells to mature into functioning effector T cells after encounter with their specific antigen. (Chapter 11)
long-lived memory T cells produced during an adaptive immune response that enter the infected non-lymphoid tissue and reside there, never to return to the circulation. (Chapter 11)
long-lived memory T cells produced during an adaptive immune response that enter the infected non-lymphoid tissue and reside there, never to return to the circulation. (Chapter 11)