Chapter 2Innate Immunity: the Immediate Response to Infection
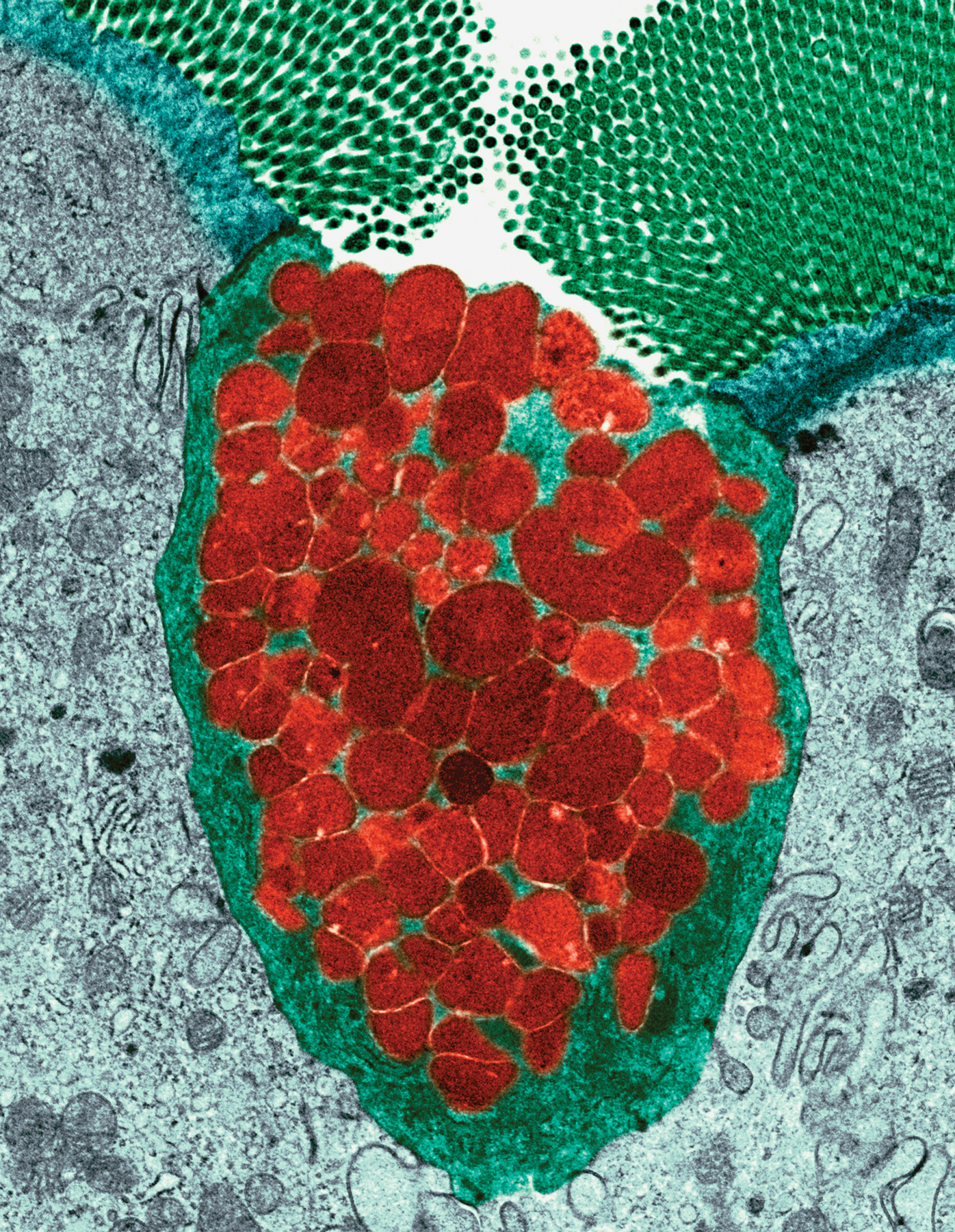
Although we become conscious of infectious agents only when suffering from the diseases they cause, microorganisms are always with us. Almost all the microorganisms we encounter are prevented from ever causing disease. To do this, the immune system has three lines of defense, which operate at different times during an infection. Of these, the first two involve innate immunity (see Section 1-4). This chapter examines the first lines of defense: the physical barriers and molecular mechanisms of innate immunity, which are always ready for action and function from the start of infection. The second line of defense comprises innate immune mechanisms that mobilize once cells of the immune system have detected the presence of infection. These induced mechanisms of innate immunity, which require from a few hours to 4 days to become fully functional, are the subject of Chapter 3. The third line of defense, the adaptive immune response, is reserved for the small minority of infections that are not subdued by innate immunity. Although slow to start, the adaptive immune response is more powerful than the innate response and, in most circumstances, has the added advantage of providing a long-lasting immunity to the pathogen. The mechanisms of adaptive immunity are covered in Chapters 4–11 of this book.
2-1Physical barriers colonized by commensal microorganisms protect against infection by pathogens
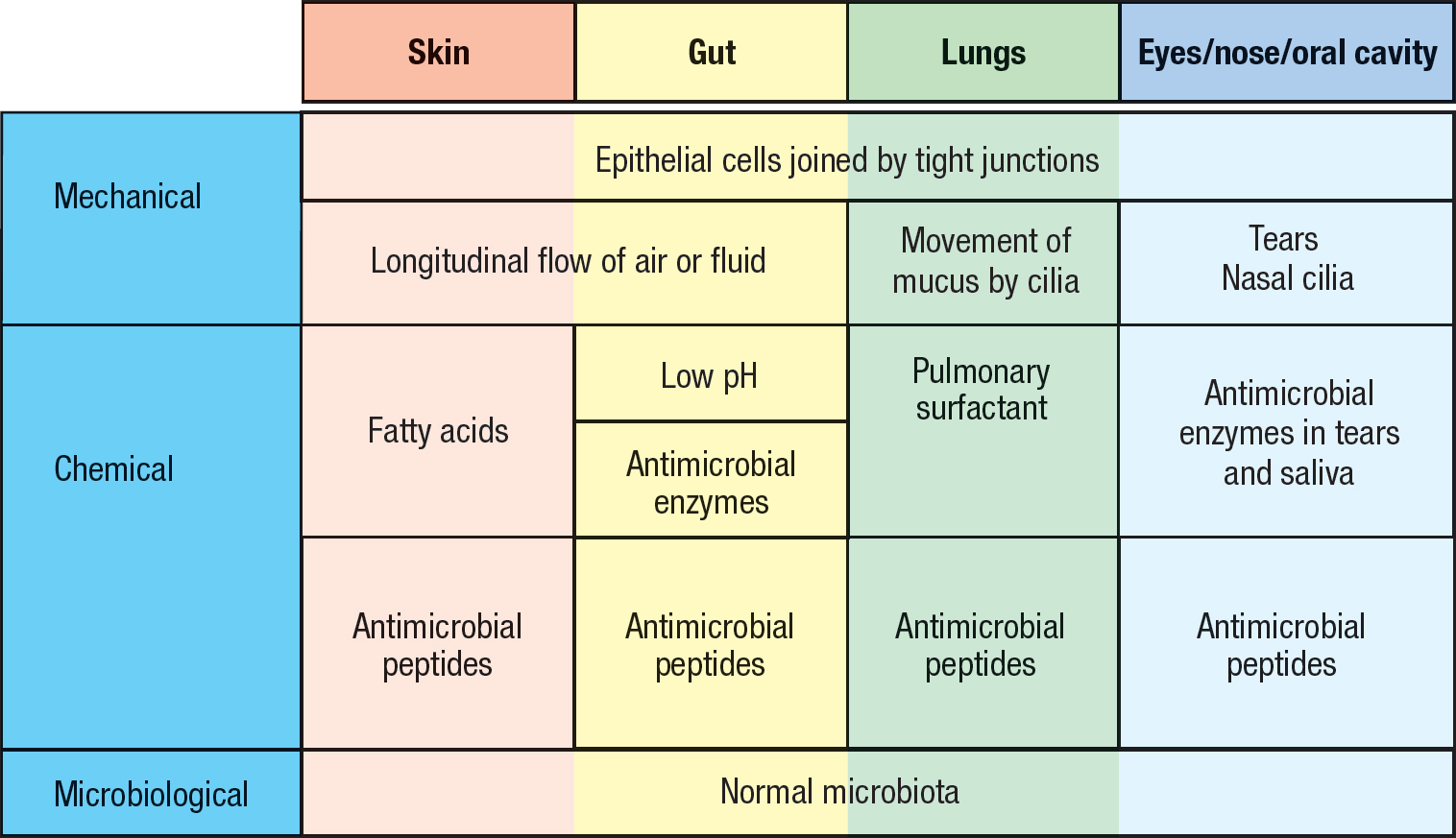
The outside surface of the human body comprises the skin and the mucosal epithelia that line the digestive, respiratory, and urogenital tracts (see Figure 1.5, p. 10). These epithelial tissues provide effective physical and chemical barriers that prevent pathogens in the external environment from gaining access to the internal tissues and organs (Figure 2.1). Further deterring pathogen invasion are the large communities of commensal microorganisms that colonize the skin and mucosal surfaces of healthy individuals (see Section 1-1). To live, multiply, and create an infection, a pathogenic microorganism must compete successfully with the resident commensals for nutrients and space. That pathogenic strains are not always competitive is exemplified by Clostridium difficile, which only takes hold in weak and unhealthy humans whose own populations of commensal bacteria have been decimated in the course of antibiotic treatment (see Figure 1.2, p. 5).
Before birth, mammalian babies have no commensal microorganisms. Starting at birth and contact with the mother’s vagina, the infant’s skin and mucosal surfaces begin to be populated by commensals acquired from family members, friends, and pets. The mucosal surfaces provide a much larger and richer habitat for commensal microorganisms than the skin; the gut is a particularly good habitat because it provides an abundant and reliable source of food. More than 1000 species of bacteria inhabit the human gut, with most of them residing in the colon. An average human harbors a total of 3.8 × 1013 bacteria, a number comparable to the 3.0 × 1013 cells of the human body. Of these cells 90% are hematopoietic cells. Having coevolved with placental mammals for more than 100 million years, commensal bacteria have developed symbiotic relationships with their human hosts that mutually improve their nutrition, metabolism, and general health. Direct evidence for the benefit of maintaining commensal relationships is the poor health of laboratory mice raised from birth in a germ-free environment. The population of commensal microorganisms, known as the microbiota, is an important and integral part of a healthy human body and one that needs to be looked after. Because the immune system is only partially developed at birth, its continuing development after birth is strongly influenced by the acquisition of the microbiota.
2-2Different immune responses are targeted to extracellular and intracellular infections
Pathogens are a diverse collection of organisms that exploit the human body in numerous ways. A key difference is between pathogens that populate the spaces between human cells to produce extracellular infections and pathogens that replicate inside human cells to produce intracellular infections (Figure 2.2). Extracellular pathogens are accessible to soluble, secreted molecules of the immune system, whereas intracellular pathogens are not. The strategy the immune system uses to attack intracellular pathogens is to kill the human cells in which the pathogens are living and replicating. This sacrifice interferes with the pathogen’s life cycle and exposes any pathogens released from the dead cells to the soluble molecules of the immune system.
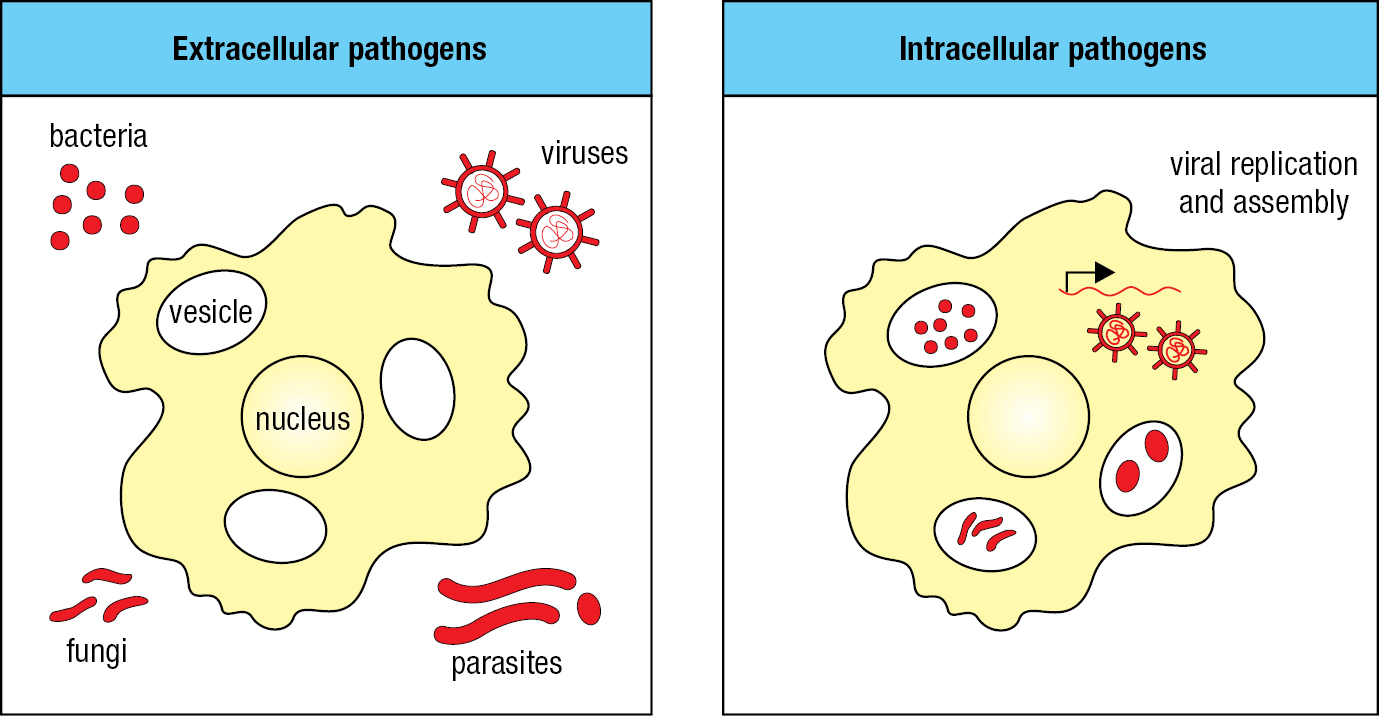
Many pathogens have life cycles that involve both extracellular and intracellular forms. For example, when particles of influenza virus first enter the respiratory tract they are extracellular and susceptible to attack by soluble molecules of the immune system. Once viral particles have infected the epithelial cells that line the respiratory tract, the virus is no longer susceptible to soluble immune-system proteins. By contrast, at this stage in the viral life cycle, the infected cells can be recognized and killed by effector cells of the immune system, which detect virus-induced changes on the surface of the infected cells. With replication of the virus in the epithelial cells and the release of viral particles into the extracellular space, the virus again becomes a target for soluble effector molecules.
2-3Complement is a system of plasma proteins that mark pathogens for destruction
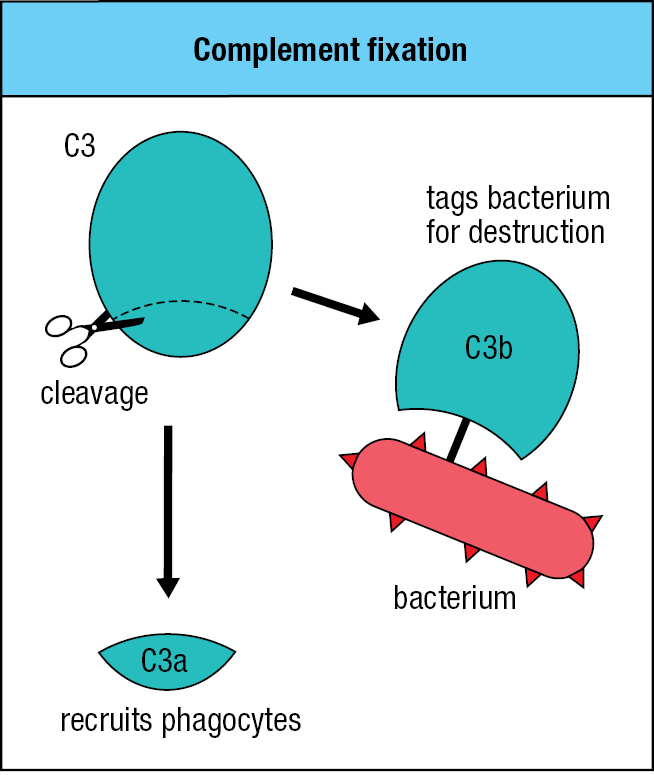
Once a pathogen penetrates an epithelial barrier and starts to live in a human tissue, the mechanisms of innate immunity come into play. One of the first weapons to fire is provided by a system of soluble proteins that are made constitutively in the liver and are present in the blood, lymph, and extracellular fluids. Collectively, these plasma proteins form the complement system, a name that is often shortened to complement. Complement coats the surface of bacteria and extracellular virus particles, making them more susceptible to phagocytosis by neutrophils and macrophages (see Section 1-4). Without a coating of complement, many bacteria resist phagocytosis, particularly those that have thick polysaccharide capsules.
Many complement components are proteolytic enzymes (proteases) that circulate in the blood, lymph, and tissues as functionally inactive forms known as zymogens. When a tissue becomes infected with a microbial pathogen, complement activation is induced and proceeds by a cascade of enzymatic reactions, in which each protease cleaves and activates the next protease in the pathway. Each protease is specific for the complement component it cleaves, and cleavage usually occurs at a single, specific site. Most of these enzymes belong to the serine protease family, which also includes the digestive enzymes chymotrypsin and trypsin.
Although more than 30 proteins make up the complement system, complement component 3 (C3) is by far the most important. People lacking any other complement component have a relatively minor immunodeficiency, but people lacking C3 are prone to successive severe infections. Activation of the complement system by infection leads to the cleavage of C3 into a small C3a fragment and a larger C3b fragment. In the process, some C3b fragments become covalently bound to the pathogen’s surface (Figure 2.3). This attachment of C3b to pathogen surfaces is the essential function of the complement system; it is called complement fixation because C3b becomes firmly fixed to the pathogen. The bound C3b tags the pathogen for phagocyte-mediated destruction and can also organize the formation of protein complexes that damage the pathogen’s membrane. The soluble C3a fragment also contributes to the immune response by acting as a chemoattractant that recruits phagocytes and other effector cells from the blood to the site of infection.
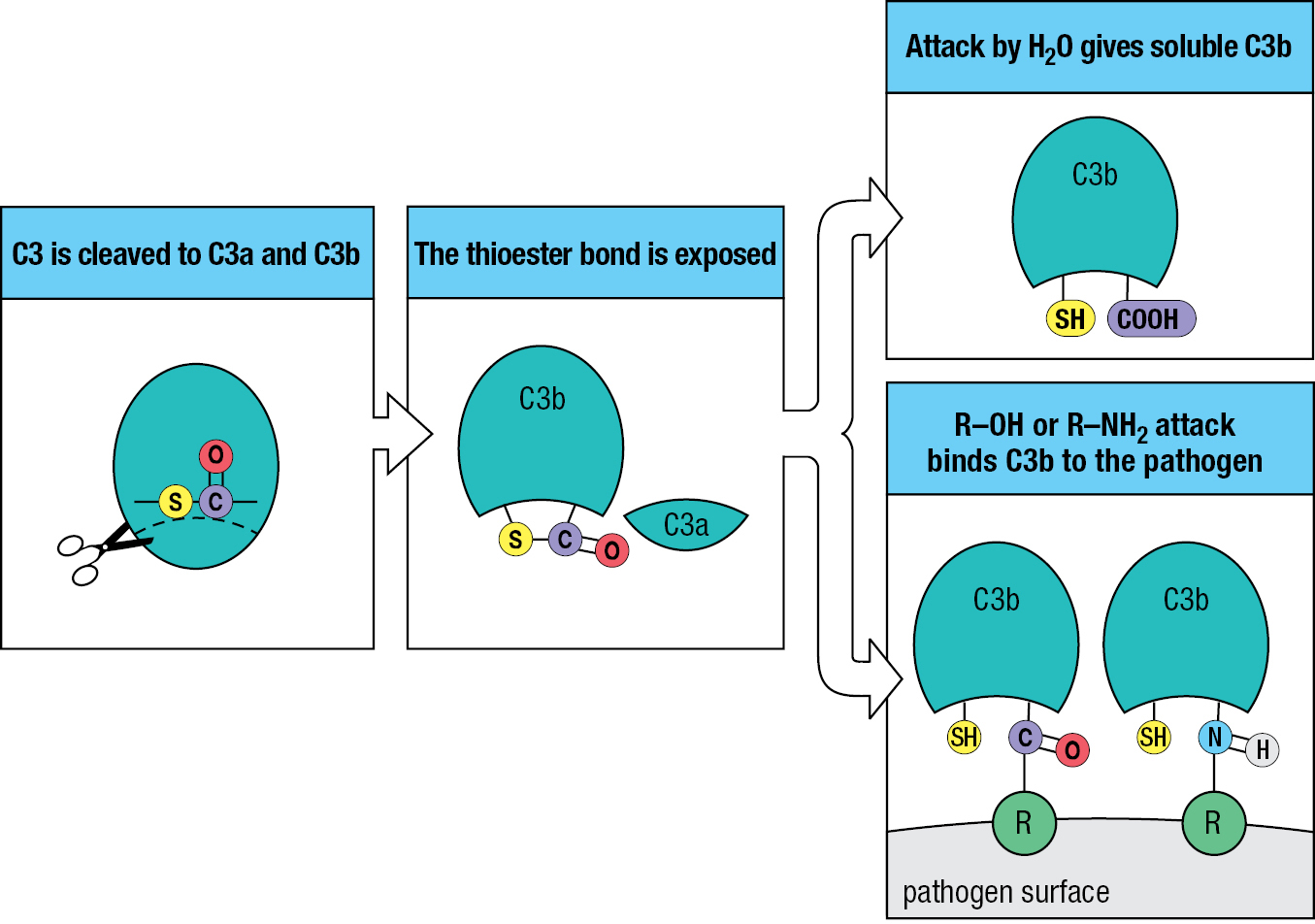
The unusual feature of C3 that underlies its unique and potent function is a high-energy thioester bond within the glycoprotein. C3 is made and enters the circulation in an inactive form, in which the thioester is sequestered and stabilized within the hydrophobic interior of the protein. When C3 is cleaved into C3a and C3b, the bond is exposed and becomes subject to nucleophilic attack by water molecules or by the amino and hydroxyl groups of proteins and carbohydrates on pathogen surfaces. This reaction causes a small fraction of the C3b to become covalently bound to the pathogen (Figure 2.4). For the vast majority of C3b molecules, water attacks their thioester bonds, causing them to stay in solution in an inactive form.
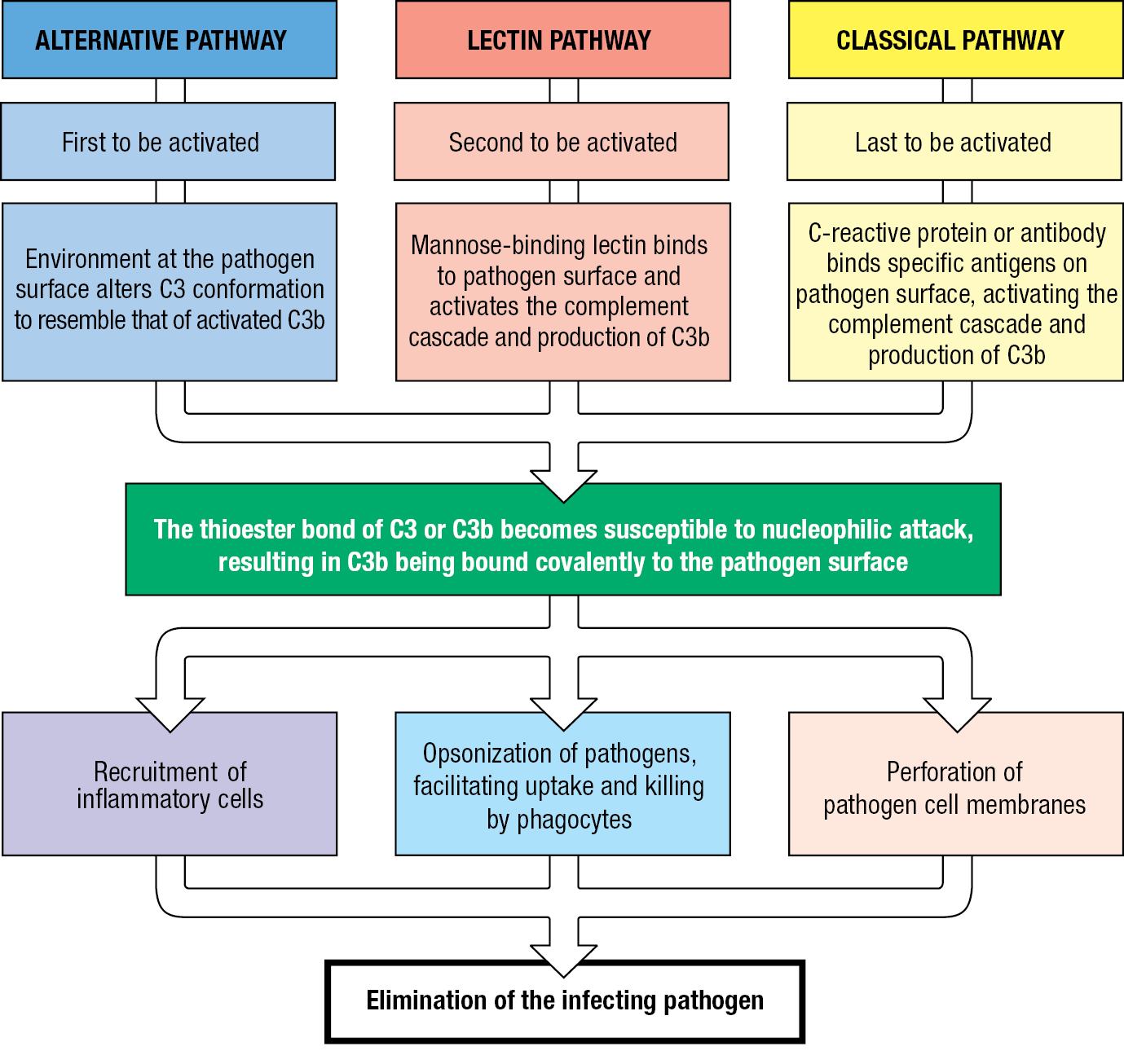
There are three pathways of complement activation. They are initiated in different ways and differ in the first few reactions of the cascade, but they all lead to C3 activation, deposition of C3b on the pathogen’s surface, and the recruitment of similar effector mechanisms for pathogen destruction (Figure 2.5). Operating from the start of infection is the alternative pathway of complement activation. The lectin pathway of complement activation also contributes to the innate immune response, but it is induced by infection and requires time to gain strength. The third pathway, the classical pathway of complement activation, contributes to both innate and adaptive immunity and requires the binding of either antibody or an innate immune-system protein called C-reactive protein to the pathogen’s surface. The names of the pathways reflect the order of their scientific discovery: the classical pathway was discovered first, then the alternative pathway, and lastly the lectin pathway. The name complement was coined because the effector functions provided by these proteins were seen to ‘complement’ the pathogen-binding function of antibodies in the classical pathway of complement activation and pathogen destruction.
2-4At the start of an infection, complement activation proceeds by the alternative pathway
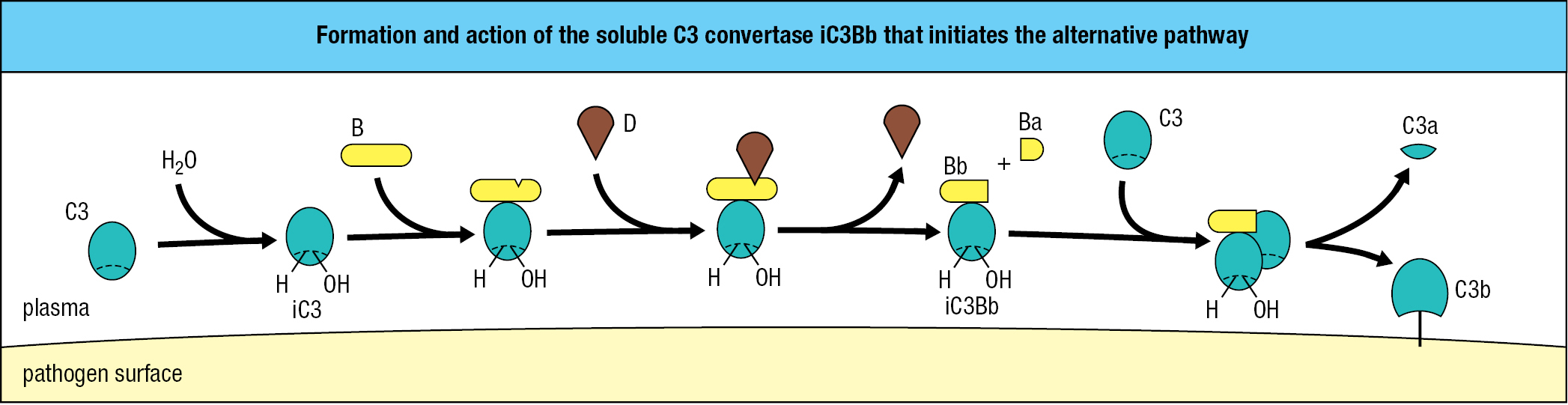
The alternative pathway of complement activation is one of the first responses of the innate immune system, especially to bacterial infection, and is the pathway described in detail in this chapter. The other pathways are described in later chapters. C3 is made in the liver and enters the circulation in a conformation that sequesters and protects its thioester bond within the hydrophobic interior of the protein. At a slow rate, and without being cleaved, C3 spontaneously changes conformation and exposes its thioester bond. In the aqueous environment of the blood, the thioester bond becomes active and quickly makes a covalent bond that attaches C3 to a nearby molecule that has either an amino or a hydroxyl group. This is almost always a water molecule, because they are so abundant, to give a form of C3 called iC3 or iC3C3(H2O). This hydrolysis, the first step in the alternative pathway of complement activation, is described as ‘tickover’ because it resembles the idling engine of a stationary automobile. Tickover occurs all the time, in both the absence and presence of infection.
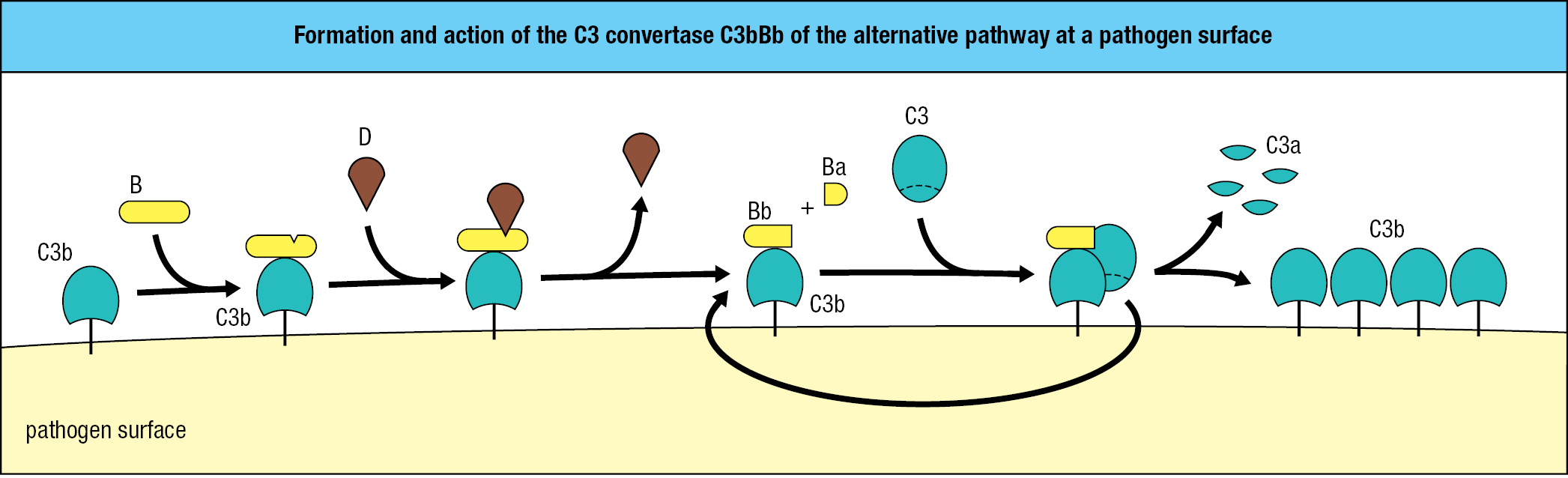
The environment near the surface of some pathogens, notably bacteria, accelerates the rate of C3 hydrolysis to iC3. Also facilitating iC3 production is the high concentration of C3 in blood (around 1.2 mg/ml). iC3 binds to the inactive form of the complement protein factor B, making factor B susceptible to cleavage by the protease factor D. This reaction produces a small fragment, Ba, which is released, and a large fragment, Bb, which has protease activity and remains bound to iC3. The iC3Bb complex is a protease that specifically cleaves C3 into the C3a and C3b fragments, exposing the thioester bond present in C3b. With C3 being activated and cleaved in large quantity, some C3b fragments become covalently attached to the amino and hydroxyl groups present on the pathogen’s outer surface (Figure 2.6).
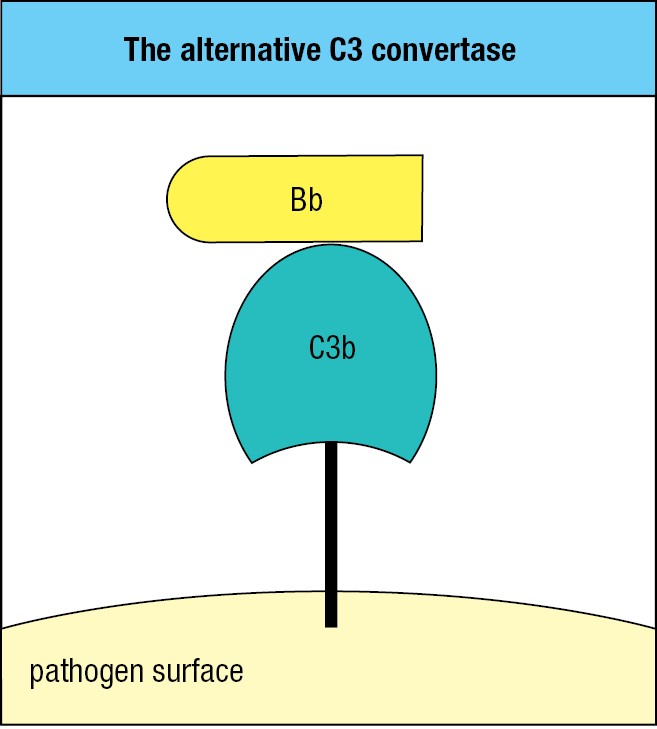
Proteases that cleave and activate C3 are called C3 convertases, iC3Bb being one example of a soluble C3 convertase. Like iC3, pathogen-bound C3b binds to factor B, which facilitates the cleavage of B by factor D. This leads to the release of Ba and formation of a C3bBb complex on the microbial surface. C3bBb, the C3 convertase of the alternative pathway, works right at the surface of the pathogen (Figure 2.7). C3bBb binds C3 and cleaves it into C3a and C3b with activation of the thioester bond. Because this convertase is present on the pathogen’s surface and cannot diffuse away like iC3Bb, it is more efficient in fixing C3b fragments to the surface of a pathogen. Once some C3 convertase has been assembled, it cleaves more C3 and fixes more C3b at the microbial surface, leading to the assembly of yet more convertase. From the initial deposition of a few molecules of C3b, the pathogen rapidly becomes coated with C3b (Figure 2.8).
2-5Regulatory proteins determine the extent and site of C3b deposition
As we saw in Section 2-4, the alternative C3 convertase, C3bBb, is capable of rapid, runaway reactions because one molecule of C3bBb catalyzes the formation of numerous additional C3bBb molecules. Two broad categories of complement control proteins evolved to regulate these reactions, which they do mainly by stabilizing or degrading C3b at cell surfaces. One category comprises plasma proteins that interact with C3b attached to human and microbial cell surfaces; the other category contains membrane proteins on human cells that prevent complement fixation at the cell surface.
The plasma protein properdin (factor P) increases complement activation by binding to C3bBb on microbial surfaces and thus preventing its degradation by proteases (Figure 2.9, top panel). Countering the effect of properdin is factor H, a plasma protein that binds C3b and facilitates its further cleavage to iC3b by factor I, a plasma serine protease (Figure 2.9, center panel). Fragment iC3b cannot assemble a C3 convertase, so the combined effect of factors H and I is to decrease the amount of C3 convertase on the pathogen’s surface.
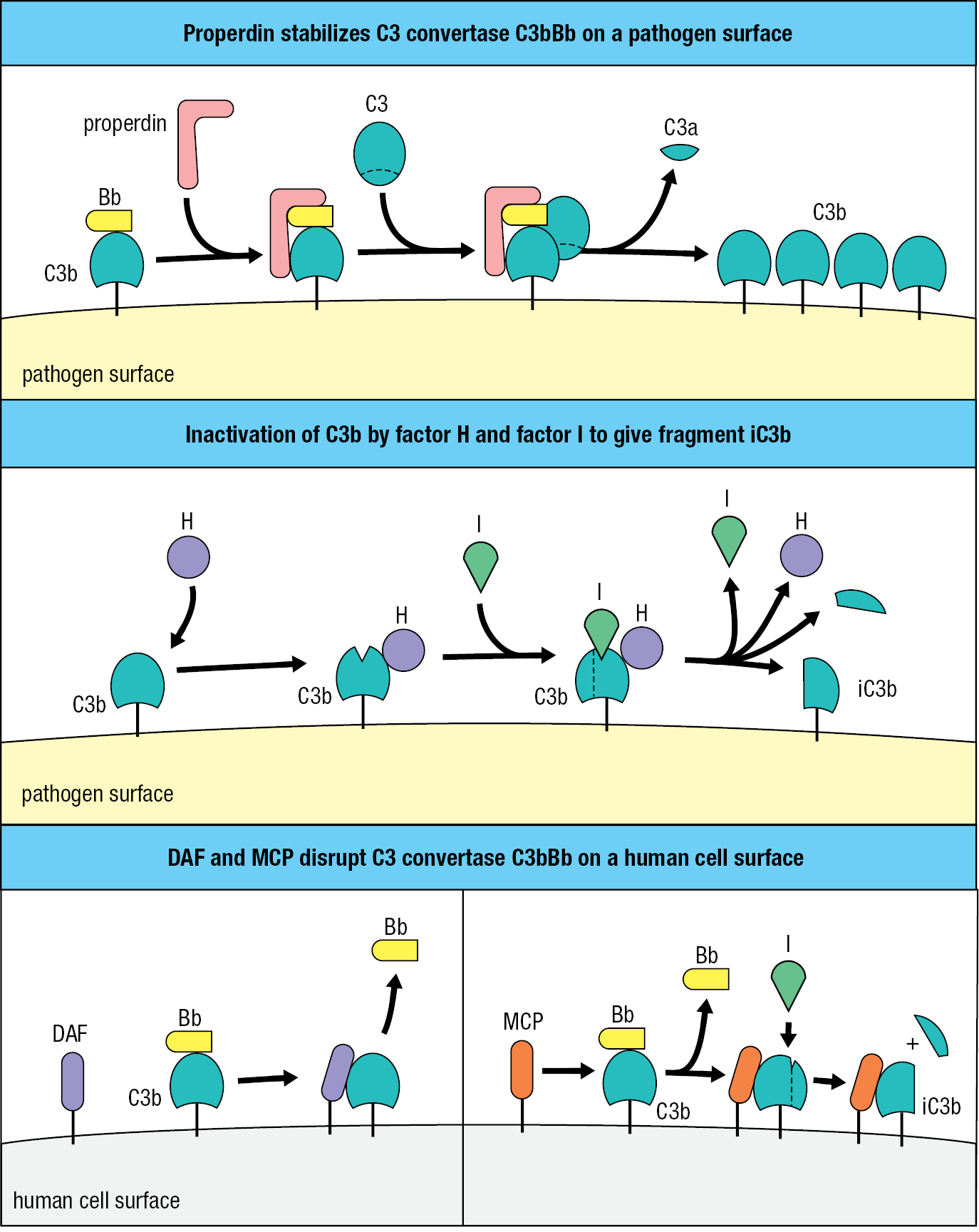
Emphasizing the importance of negative regulation by factors H and I is the immunodeficiency suffered by people who lack factor I. In these individuals, formation of the C3 convertase C3bBb runs away unchecked until the C3 reservoir in blood, extracellular fluid, and lymph is exhausted. When faced with bacterial infections, individuals with factor I deficiency fix very little C3b to bacterial surfaces, causing inadequate clearance of bacteria by phagocytes. Consequently, these individuals are particularly susceptible to ear infections and abscesses caused by encapsulated bacteria. By contrast, healthy individuals control these bacteria by coating their thick polysaccharide capsules with complement, thus ensuring their efficient clearance by neutrophils.
The second category of complement control proteins comprises membrane proteins of human cells that interfere with complement activation at human cell surfaces. Decay-accelerating factor (DAF) binds to the C3b component of the alternative C3 convertase, causing its dissociation and inactivation. Membrane cofactor protein (MCP) also has this function, but the binding of MCP to C3b makes C3b also susceptible to cleavage and inactivation by factor I (Figure 2.9, bottom panel). The functions of MCP are similar to those of the soluble complement regulator, factor H, which can also be membrane-associated; factor H has a binding site for sialic acid, a component of human cell-surface carbohydrates that is not made by most bacteria. As a strategy to evade the actions of complement, some species of bacteria, such as Streptococcus pyogenes and Staphylococcus aureus, cover their cell surfaces with sialic acid and so mimic human cells. Consequently, when C3b becomes deposited on the surface of these bacteria, it is readily inactivated by factor H bound to the bacterial sialic acid.
Many of the diverse proteins that regulate complement, such as DAF, MCP, and factor H, are elongated structures built from structurally similar elements known as complement control protein (CCP) modules. Each module consists of about 60 amino acids that fold into a compact sandwich formed from two slices of β-pleated sheet stabilized by two conserved disulfide bonds that cross-link the two sheets. Proteins made up of CCP modules are also called regulators of complement activation.
The combined effect of the reactions that promote and regulate C3 activation is to ensure that C3b is deposited only on the surfaces of pathogenic microorganisms and not on human cells. In this manner, the complement system provides a simple and effective way of distinguishing human cells from microbial cells and for guiding lethal mechanisms toward invading pathogens and away from healthy cells and tissues.
2-6The macrophage is a first line of cellular defense against an invading microorganism
When a pathogen invades a human tissue, the first effector cells of the immune system it encounters are the resident macrophages (see Section 1-6). Macrophages are prevalent in the connective tissues, the linings of the gastrointestinal and respiratory tracts, the liver, and the alveoli of the lungs. Liver macrophages are also known as Kupffer cells. Macrophages are long-lived phagocytic cells that contribute to both innate and adaptive immunity.
Although macrophages can phagocytose bacteria and other microorganisms in a nonspecific fashion, their efficiency is improved by macrophage cell-surface receptors that engage specific ligands on the microbial surface. One such receptor, complement receptor 1 (CR1), binds C3b fragments that were deposited on a pathogen’s surface during activation of the alternative pathway (see Figure 2.8). Interaction of the arrayed C3b fragments on a pathogen with CR1 arrayed on the macrophage facilitates the engulfment and degradation of the pathogen. This process, by which a coating of C3b improves the efficiency of phagocytosis, is known as opsonization (Figure 2.10).
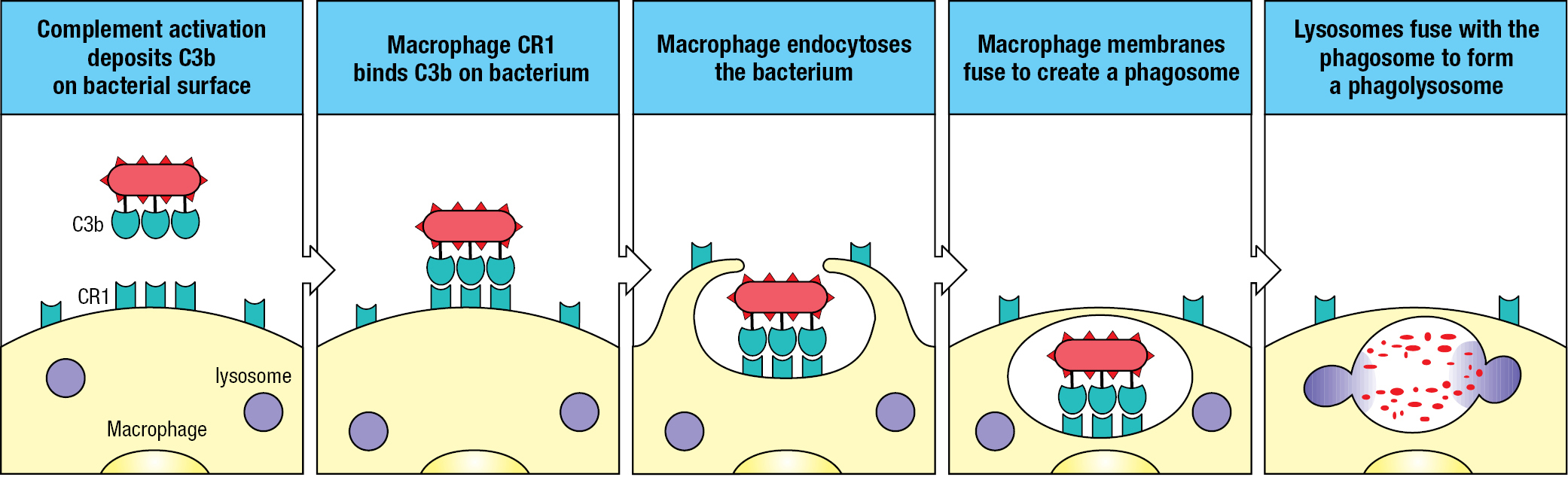
CR1 also acts to protect the surface of cells on which it is expressed. Like MCP and factor H, CR1 disrupts the C3 convertase by making C3b susceptible to cleavage by factor I. During phagocytosis, some of a macrophage’s CR1 molecules have this protective role, whereas others engage the C3b fragments deposited on the pathogen’s surface. Like MCP and factor H, CR1 is made up of CCP modules.
Two other macrophage receptors, complement receptor 3 (CR3) and complement receptor 4 (CR4), bind iC3b fragments on microbial surfaces. Although the iC3b fragment has no C3 convertase activity, it facilitates phagocytosis and pathogen destruction as a ligand for CR3 and CR4. These receptors, which are not made up of CCP modules, are examples of integrins, a large family of cell-surface glycoproteins that contributes to adhesive interactions between cells. In the phagocytosis of complement-coated pathogens, the combination of CR1, CR3, and CR4 is much more effective than any single complement receptor.
From the beginning of infection, the alternative pathway deposits C3b on a pathogen’s surface. These molecular tags improve the capacity and speed of macrophages to recognize, swallow, and digest the invading pathogen.
2-7The terminal complement components make pores in microbial membranes
The terminal components of the complement cascade comprise the C5, C6, C7, C8, and C9 proteins (Figure 2.11). These five components cooperate to form the membrane-attack complex (MAC), a large pore that is assembled in the membrane of bacterial and eukaryotic pathogens and kills them by perturbing their structural integrity.
| Five terminal complement proteins form the membrane-attack complex | ||
|---|---|---|
| Protein | Level in serum (μg/ml) | Function |
| C5 | 85 | On activation by cleavage the soluble C5b fragment initiates assembly of the membrane-attack complex (MAC) in solution |
| C6 | 60 | C6 binds to and stabilizes C5b, thereby forming a binding site for C7 |
| C7 | 55 | C7 binds to C5b6 and exposes a hydrophobic region that permits attachment to the cell membrane |
| C8 | 55 | C8 binds to C5b67, exposing a hydrophobic region that inserts into the cell membrane |
| C9 | 60 | Polymerization of C9 on the C5b678 complex forms a membrane-spanning channel that disrupts the cell’s integrity and can cause cell death |
C5 is structurally similar to C3 but lacks a thioester bond and has a different function. C5 is activated by the alternative C5 convertase, which consists of two C3b fragments and one Bb fragment and is designated C3b2Bb. This convertase cleaves C5 into a smaller C5a fragment, similar to C3a, and a larger C5b fragment, similar to C3b (Figure 2.12).
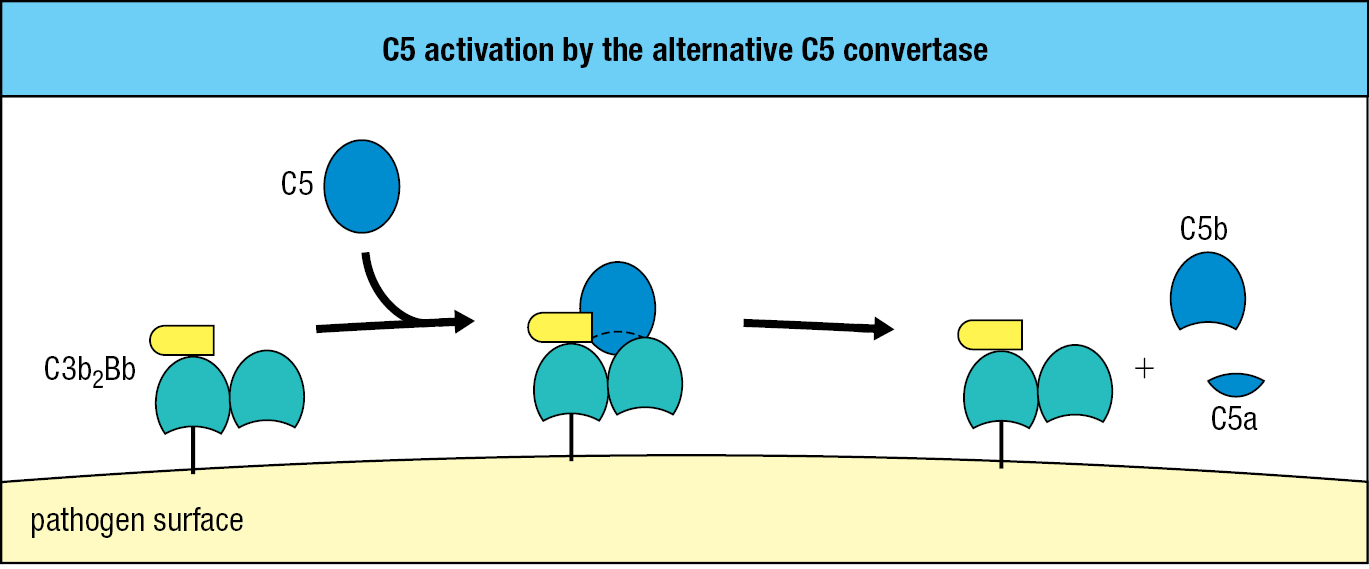
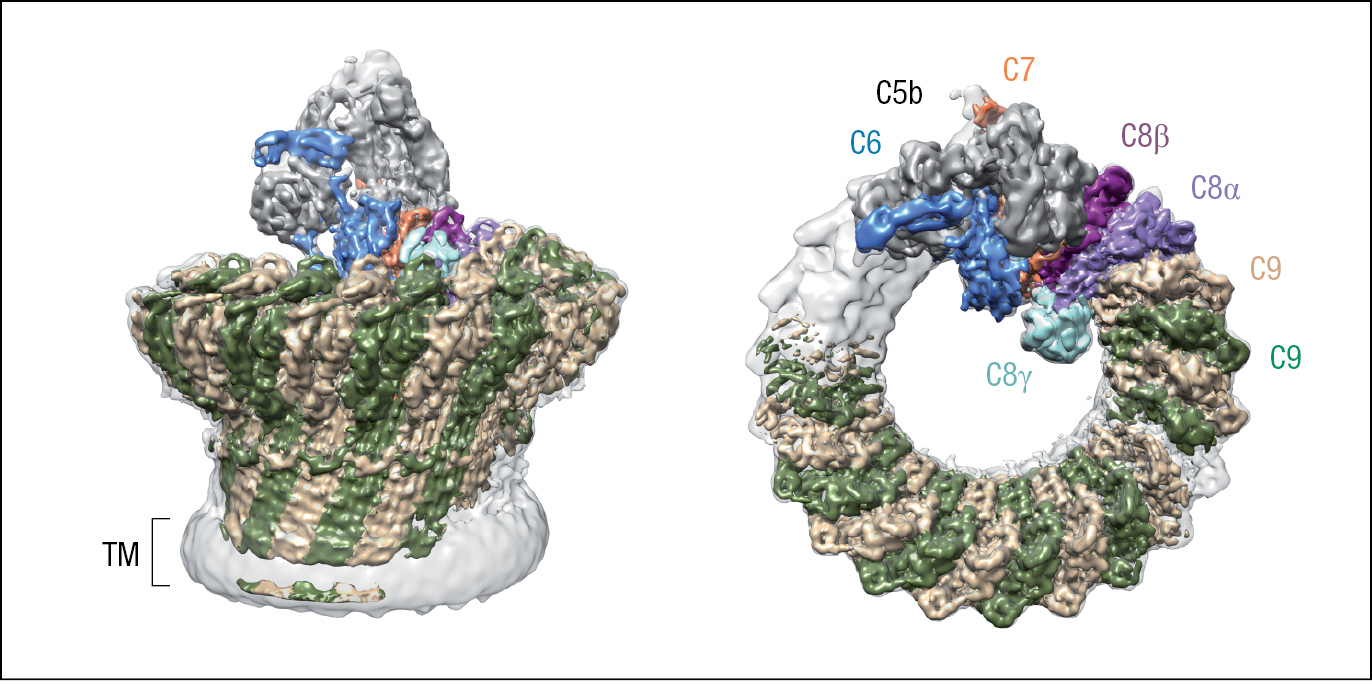
The function of C5b is to initiate MAC formation. In succession, C6 and C7 bind to C5b. These interactions expose a hydrophobic region of C7, which then inserts into the lipid bilayer. Likewise, when C8 binds C5b, a hydrophobic site of C8 is exposed and inserted into the membrane. These events initiate polymerization of C9 and MAC formation. The functional complex consists of one molecule each of C5b, C6, and C7, three molecules of C8, and 18 molecules of C9 that form a large pore with a diameter of 100 Å (10 nm) (Figure 2.13). The structure of the MAC obtained by cryoelectron microscopy is shown in Figure 2.14.
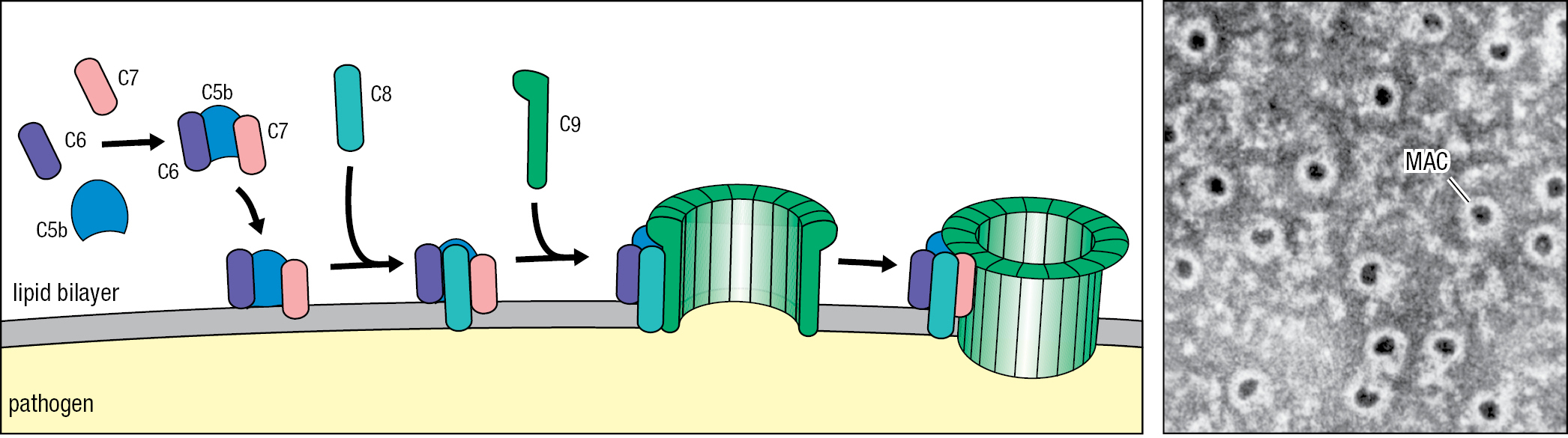
Both soluble and membrane-bound proteins protect healthy human cells from the damaging effects of complement activation. S protein, clusterin, and factor J are soluble proteins that prevent association of the complex of C5b, C6, and C7 with cell membranes. Present on human cell surfaces, homologous restriction factor (HRF) and CD59 (also called protectin) are proteins that prevent C9 recruitment to the complex of C5b, C6, C7, and C8 (Figure 2.15). A shared feature of DAF, HRF, and CD59 is their attachment to the plasma membrane by a glycosylphosphatidylinositol lipid tail. Impaired synthesis of the tail causes the human disease paroxysmal nocturnal hemoglobinuria (PNH). Characterizing this immunodeficiency are episodes of complement-mediated lysis of erythrocytes, which have no DAF, HRF, or CD59 on their surfaces and so are not protected from the actions of complement. An effective treatment for PNH is a humanized monoclonal antibody (eculizumab) that is specific for C5 and prevents its cleavage and activation by C5 convertases.

The most notable effect of a deficiency in the terminal components of complement is increased susceptibility to infection by bacteria of the genus Neisseria, which cause gonorrhea and a common form of bacterial meningitis.
2-8Small peptides released during complement activation induce local inflammation
In the course of complement activation, C3 and C5 are both cleaved into two fragments. Of these, the larger C3b and C5b fragments continue the complement cascade, whereas the smaller C3a and C5a fragments are ligands for receptors on phagocytes, endothelial cells, and mast cells. These ligand–receptor interactions increase inflammation at the site of complement activation. Inflammation (see Section 1-4) is a major consequence of the innate immune response to infection, which is also known as the inflammatory response. In some circumstances, C3a and C5a induce anaphylactic shock, an acute and powerful inflammatory reaction that affects tissues throughout the body and can be lethal. In this context, C3a and C5a are described as anaphylatoxins, with C5a being more potent and stable than C3a. The C3a and C5a receptors are structurally related, and of a type that is embedded in cell membranes and signals by activating an intracellular guanine nucleotide–binding protein.
Anaphylatoxins induce contraction of smooth muscle and degranulation of mast cells and basophils. These cells release histamine and other vasoactive substances that increase capillary permeability. Anaphylatoxins affect local blood vessels by increasing their vascular permeability and blood flow. These changes facilitate the exit of plasma proteins and cells from the blood and their passage to sites of infection in the tissues (Figure 2.16).
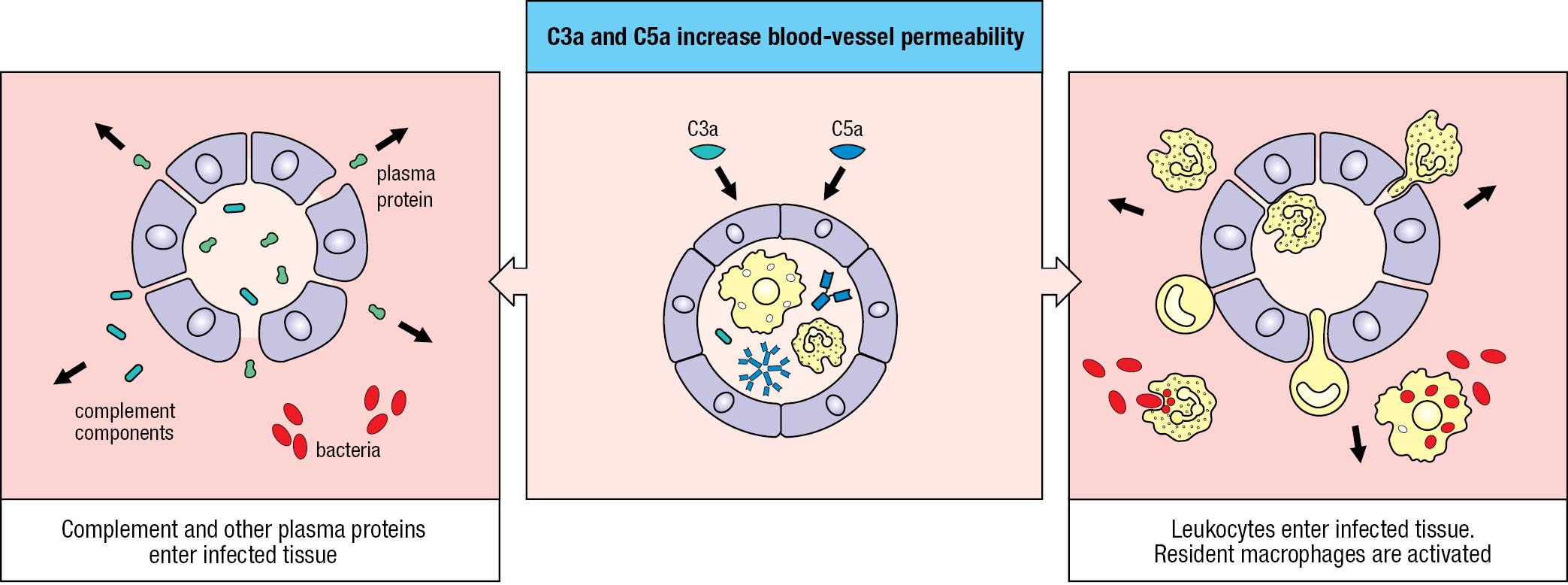
C5a induces neutrophils and monocytes to adhere strongly to blood vessel walls, and as a chemoattractant it commands these phagocytes to migrate toward sites of complement fixation. In addition, C5a increases their phagocytic capacity and the levels of CR1 and CR3 on their surfaces. By these means, the anaphylatoxins act in concert with other complement components to speed the destruction of pathogens by phagocytes.
2-9Several systems of plasma proteins limit the spread of infection
In countering the invasion and colonization of human tissues by pathogenic microorganisms, the complement cascade cooperates with other systems of plasma proteins. Blood vessels damaged by pathogens activate the coagulation system, a cascade of enzymes in plasma that cooperates with platelets to form blood clots. Immobilized in the clots, pathogens cannot enter the blood and lymph and be transported throughout the body. Clotting also serves to decrease blood and fluid loss. During clot formation the platelets degranulate, releasing a variety of highly active agents. These include prostaglandins, hydrolytic enzymes, and growth factors that contribute to the recruitment of immune-system cells, antimicrobial defenses, and tissue repair. The kinin system, another enzymatic cascade of plasma proteins, is induced by damaged tissue and leads to the production of bradykinin. This nonamer peptide reduces hypertension, dilates blood vessels, and relaxes smooth muscle in the damaged tissue. These properties facilitate the elimination of invading pathogens and repair of damaged tissue.
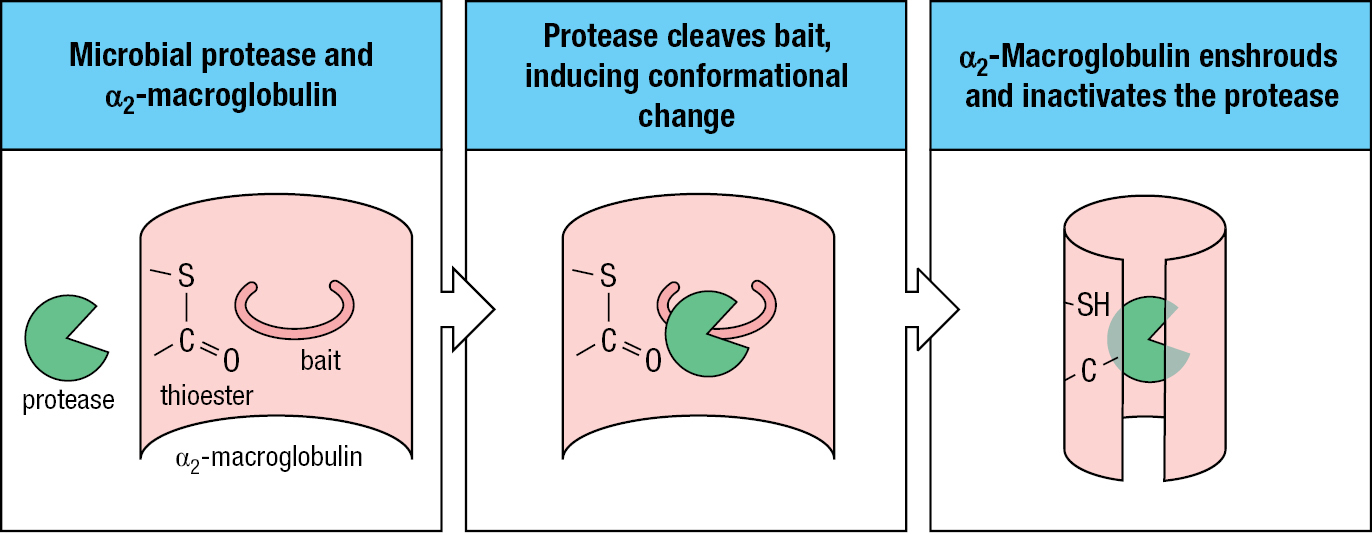
In the course of invasion, many pathogens make or acquire proteases that inactivate or subvert antimicrobial proteins and degrade human tissues. For example, the bacterium Streptococcus pyogenes acquires a coat of human plasmin on its outer surface, which makes it invisible to the immune system. In response, human secretions and plasma contain protease inhibitors, which constitute about one-tenth of the serum proteins. Among these are the α2-macroglobulins, large 180-kDa glycoproteins that circulate as monomers, dimers, and trimers and inhibit a broad range of proteases. Structurally, the α2-macroglobulins resemble C3 and have a thioester bond. The α2-macroglobulin can lure a microbial protease with its ‘bait’ region, which the protease will cleave. This activates the thioester, which covalently links the protease to the α2-macroglobulin. In turn this induces a conformational change in the α2-macroglobulin, which envelops the protease and forms a complex that is quickly bound by a specific receptor on macrophages, hepatocytes, and fibroblasts (Figure 2.17).
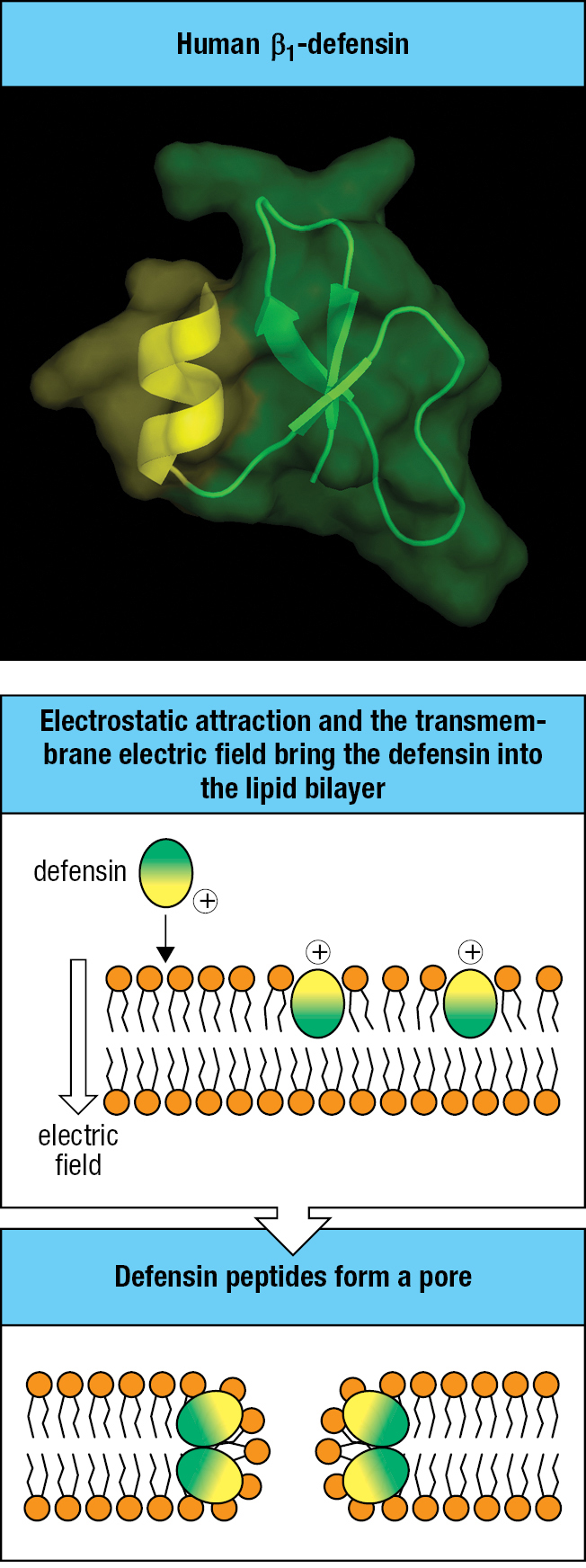
2-10Defensins are antimicrobial peptides that kill pathogens by disrupting their membranes
Antimicrobial peptides are a major component of the innate immune response to infection. The predominant family of human antimicrobial peptides is the defensins, which exhibit a broad range of antimicrobial and immunomodulatory functions. In particular, defensins are the only components of the innate immune response that are known to neutralize a broad range of structurally diverse microbial toxins. These toxic bacterial proteins, such as those that cause cholera and diphtheria, include some of the deadliest natural products on Earth.
Defensins are peptides of 30–40 amino acids that are rich in arginine residues and have a common structure, consisting of a three-stranded antiparallel β sheet stabilized by three intrachain disulfide bonds. They are of two classes with different structure and divergent evolution—the α-defensins and the β-defensins. The defensin molecule is amphipathic in character, meaning that its surface has both hydrophobic and hydrophilic regions. This property allows defensins to penetrate microbial membranes, disrupt their integrity, and cause lysis—the mechanism by which they destroy bacteria, fungi, and enveloped viruses (Figure 2.18).
Common features of bacterial toxins exploited by defensins are their structural plasticity and thermodynamic instability. These properties, which are essential for the toxic functions, distinguish the toxins from almost all human proteins. When a defensin binds to a microbial toxin, the defensin promotes a localized unfolding of the toxin, which destabilizes its three-dimensional structure. This has the effect of making the toxin susceptible to attack by human proteases and to physical denaturation and precipitation. By this general mechanism, one type of defensin can eliminate the toxins made by many different microbial pathogens, and which vary widely in their protein sequence. Proteins that facilitate the correct folding of other proteins are called chaperones. The defensins are sometimes called anti-chaperones because they promote protein unfolding and denaturation (Figure 2.19).
| Bacterial toxins neutralized by defensins | ||||
|---|---|---|---|---|
| Toxin | Defensin | Class | Mechanism | Biological effect |
| Bacillus anthracis lethal factor | HNP1,2,3 | α | Enzyme | Neutralizes lethal factor |
| Diphtheria toxin, Pseudomonas exotoxin A | HNP1,2,3 | α | Enzyme | Inhibit toxins’ enzymatic activity |
| Clostridium difficile toxin | HNP1,3 HD5 | α | Enzyme | Inhibit toxin’s enzymatic activity |
| Cholesterol-dependent cytolysins | HNP1,3 HD5 | α | Pore formation | Inhibit cytolysins |
| Vibrio cholerae, Aeromonas hydrophila toxins | HNP1, HD5 | α | Enzyme | Inhibit toxins’ actin crosslinking and cysteine protease domains |
| Staphylococcus aureus Panton–Valentine leukocidin | HNP3 | α | Pore formation | Decreases pore formation and prevents neutrophil lysis |
| Neisseria gonorrhoeae toxins | LBD1,2,4 | β | Enzyme | Inhibit auto-ADP-ribosyltransferase |
| B. anthracis lethal factor | hBD3 | β | Enzyme | Neutralizes lethal factor |
Four of the six α-defensins are components of neutrophil granules and are named human neutrophil proteins (HNP) 1–4. Also known as myeloid defensins, they participate in the innate immune response in infected tissues and organs. The other two α-defensins—human defensins 5 and 6 (HD5 and HD6)—are enteric defensins secreted by Paneth cells of the intestinal epithelium that provide defense against infections of the gut mucosa. The β-defensins are more numerous and less studied than the α-defensins. Of more than 30 human β-defensin genes, protein products have been identified for less than one-half of them.
2-11Pentraxins are plasma proteins that bind microorganisms and deliver them to phagocytes
The pentraxins are a family of cyclic multimeric proteins that circulate in the blood and lymph, bind to the surface of various pathogens, and target them for destruction. Defining the pentraxins is a characteristic 200-residue pentraxin domain at the carboxy-terminal end of the polypeptide. Two subfamilies of pentraxins are distinguished. The short pentraxins are made by hepatocytes in the liver and are represented by serum amyloid P component (SAP). The long pentraxins, represented by PTX3, are made by a variety of cells, including myeloid, endothelial, and epithelial cells, but not by liver hepatocytes (Figure 2.20). The pentraxins function as bridging molecules: they bind pathogens with one binding site and use a second site to bind to human cell-surface receptors, for example CD89 on phagocytes. When the cell-surface receptors are cross-linked by the pentraxin-coated pathogen, the phagocyte is signaled to engulf and destroy the pathogen. The pentraxins have a similar role in the innate immune response to that of antibodies in the adaptive immune response, and both pentraxins and antibodies bind to the same surface receptors on phagocytes, such as CD89, which is a cell-surface receptor for the IgA class of antibodies.
| Type | Name | Source | Ligands |
|---|---|---|---|
| Short pentraxin | Serum amyloid P component | Liver hepatocytes | Bacteria, viruses, fungi, parasites |
| Long pentraxin | PTX3 | Monocytes Macrophages Dendritic cells Endothelial cells Epithelial cells | Bacteria, viruses, fungi |
Glossary
- innate immunity
- the host defense mechanisms that act from the start of an infection and do not adapt to a particular pathogen or generate immunological memory. See also induced innate immune response. (Chapter 1, Chapter 2)
- commensal microorganisms
- a microorganism that habitually lives on or in the human body; one that normally causes no disease or harm and can be beneficial. (Chapter 1, Chapter 2)
- microbiota
- the microorganisms that habitually live in or on the human body; they normally do not cause disease and in many cases provide positive benefits for human health. (Chapter 1, Chapter 2)
- extracellular infections
- invasion of the body by pathogens that live outside cells in extracellular spaces, on the surfaces of epithelia, or in the blood. (Chapter 2)
- intracellular infections
- invasion of the body by a pathogen that can replicate inside human cells. (Chapter 1)
- complement system
- a system of some 30 soluble and cell-surface proteins, comprising the complement proteins and the complement control proteins. It provides a major mechanism in innate and adaptive immunity for identifying and eliminating pathogens and their products. See also complement. (Chapter 2)
- complement
- collection of plasma proteins that act in a cascade of reactions to attack extracellular forms of pathogens in extracellular spaces and the blood. Pathogens become coated with complement proteins, which can either kill the pathogen directly or facilitate its engulfment and destruction by phagocytes. It is involved in both innate and adaptive immunity and is activated either directly or indirectly by the presence of infection. (Chapter 1, Chapter 2)
- zymogens
- the functionally inactive form of a protease, which requires cleavage by another protease to become active. (Chapter 2)
- complement activation
- the initiation of a series of reactions involving the complement proteins present in plasma and extracellular fluid, and leading to the death and elimination of the pathogen. It can be triggered either directly or indirectly by the presence of a pathogen. See also alternative pathway of complement activation; classical pathway of complement activation; complement; lectin pathway of complement activation. (Chapter 2)
- complement component 3 (C3)
- the central and most important protein of the complement system, which is cleaved into C3a and C3b in the complement reactions. C3b becomes bound covalently to pathogen surfaces and acts as an opsonin, facilitating the phagocytosis of the pathogen. C3a is an anaphylatoxin. (Chapter 2)
- complement fixation
- the covalent attachment of C3b or C4b to pathogen surfaces, which is a central feature of the action of complement because it facilitates phagocytosis of the pathogen. (Chapter 2)
- alternative pathway of complement activation
- one of three pathways of complement activation. It is triggered by the presence of infection but does not involve antibody. The early stages leading to cleavage of C3 involve iC3b, factor B, and factor D. See also classical pathway of complement activation; lectin pathway of complement activation. (Chapter 2)
- lectin pathway of complement activation
- one of the three pathways of complement activation. It is activated by the binding of a mannose-binding lectin present in blood plasma to mannose-containing peptidoglycans on bacterial surfaces. See also alternative pathway of complement activation; classical pathway of complement activation. (Chapter 2, Chapter 3)
- classical pathway of complement activation
- one of three pathways of complement activation. It is activated either by C-reactive protein or by antibody bound to antigen, and involves complement components 1, 4, and 2 in the generation of the classical C3 and C5 convertases. See also alternative pathway of complement activation; lectin pathway of complement activation. (Chapter 2)
- iC3
- the product formed when the thioester bond of complement component 3 is hydrolyzed by water. Also called
- ic3-1
- the product formed when the thioester bond of complement component 3 is hydrolyzed by water. Also called
- factor B
- plasma protein of the alternative pathway of complement. It binds to C3(H2O) or C3b and is cleaved to form part of the alternative C3 convertases (iC3Bb and C3bBb). (Chapter 2)
- factor D
- protease that cleaves factor B to Bb and Ba in the alternative pathway of complement activation. (Chapter 2)
- C3 convertases
- any of the proteolytic enzymes that are formed during complement activation and cleave complement component 3 to C3b and C3a, thereby enabling C3b to bond covalently to antigens. See also C3 convertase of the alternative pathway; classical C3 convertase. (Chapter 2)
- C3 convertase of the alternative pathway
- the alternative C3 convertase. The soluble alternative C3 convertase is composed of iC3 bound to proteolytically active Bb (iC3Bb). The membrane-bound alternative C3 convertase is composed of C3b bound to Bb (C3bBb). Both cleave C3 into C3a and C3b. (Chapter 2)
- complement control proteins
- any of a diverse group of proteins that inhibit complement activation at various stages and by different mechanisms. See also C1 inhibitor (C1INH); decay-accelerating factor (DAF); factor I; membrane cofactor protein (MCP); protectin. (Chapter 2)
- properdin (factor P)
- a plasma protein that helps to activate the alternative pathway of complement. It binds to and stabilizes the alternative pathway C3 and C5 convertases on the surfaces of bacterial cells. Also called factor P. (Chapter 2)
- factor H
- complement regulatory protein in plasma that inactivates the C3 convertase of the alternative pathway and C5 convertases by binding to C3b and rendering it susceptible to cleavage by factor I to produce inactive iC3b. (Chapter 2)
- factor I
- protease that regulates complement action by cleaving C3b and C4b into inactive forms. (Chapter 2)
- decay-accelerating factor (DAF)
- cell-surface protein that prevents complement activation on human cells. DAF binds to C3 convertases of both the alternative and classical pathways of complement activation and, by displacing Bb and C2a, respectively, prevents their action. (Chapter 2)
- membrane cofactor protein (MCP) modules
- membrane-associated complement regulatory protein on human cells that promotes the inactivation of C3b and C4b by factor I. (Chapter 2)
- complement control protein (CCP) modules
- family of structurally similar protein modules found in many of the proteins that regulate complement activity. (Chapter 2)
- regulators of complement activation
- any of a group of proteins that regulate the activity of complement and contain one or more copies of a particular structural motif of ~60 amino acid residues called the CCP (complement control protein) motif. The CCP motif is also called a sushi domain because of its similarity in shape to a slice of a sushi roll. (Chapter 2)
- Kupffer cells
- phagocytic cell in the liver that lines the hepatic sinusoids. (Chapter 2)
- complement receptor 1 (CR1)
- receptor present on macrophages and other cells that binds the C3b fragment of complement deposited on a pathogen surface, thus aiding phagocytosis of the pathogen. (Chapter 2)
- opsonization
- the coating of the surface of a pathogen or other particle with any molecule that makes it more readily ingested by phagocytes. Antibody and complement opsonize extracellular bacteria for phagocytosis by neutrophils and macrophages because the phagocytic cells carry receptors for these molecules. (Chapter 1, Chapter 2)
- complement receptor 3 (CR3)
- receptor present on macrophages and other cells that binds the iC3b fragment of complement if present on a pathogen surface, thus aiding phagocytosis of the pathogen. It is a member of the integrin family. (Chapter 2)
- complement receptor 4 (CR4)
- receptor present on macrophages and other cells that binds the iC3b fragment of complement if present on a pathogen surface, thus aiding phagocytosis of the pathogen. Like CR3, it is a member of the integrin family. (Chapter 2)
- membrane-attack complex (MAC)
- the complex of terminal complement components that forms a pore in the membrane of the target cell, damaging the membrane and leading to cell lysis and death. (Chapter 2)
- alternative C5 convertase
- the C5 convertase of the alternative pathway of complement activation. It is composed of two molecules of C3b bound to Bb (C3b2Bb) and cleaves C5 into C5a and C5b. (Chapter 2)
- S protein
- soluble regulatory protein of complement that prevents the soluble complex of C5b with C6 and C7 from associating with cell membranes. (Chapter 2)
- clusterin
- complement-regulatory protein that prevents the soluble complex of C5b with C6 and C7 from associating with cell membranes. (Chapter 2)
- factor J
- soluble complement control protein that prevents the complex of C5b with C6 and C7 from associating with host-cell membranes and initiating an attack on them. (Chapter 2)
- homologous restriction factor (HRF)
- cell-surface complement control protein that prevents the recruitment of C9 by the complex of C5b, C6, C7, and C8 and formation of the membrane-attack complex. (Chapter 2)
- CD59
- alternative name for protectin, a complement control protein. (Chapter 2)
- protectin
- a protein on the surface of human cells that prevents assembly of the complement membrane-attack complex on cell surfaces, thus protecting human cells from complement-mediated lysis. Also called CD59. (Chapter 2)
- paroxysmal nocturnal hemoglobinuria (PNH)
- a genetic disease in which the complement regulatory proteins CD59 and DAF are defective, so that complement activation leads to episodes of spontaneous hemolysis. The defect is in the attachment of CD59 and DAF to cell membranes by a glycolipid anchor. (Chapter 2)
- anaphylatoxins
- general name for complement fragments C3a and C5a, which are produced during complement activation. They induce inflammation, recruiting fluid and inflammatory cells to sites of antigen deposition. In some circumstances, C3a and C5a can induce anaphylactic reactions. (Chapter 2)
- coagulation system
- collection of enzymes and other proteins in blood that function to form blood clots. The coagulation system is activated by damage to blood vessels. (Chapter 2)
- kinin system
- enzymatic cascade of plasma proteins that is triggered by tissue damage and helps to facilitate wound healing. (Chapter 2)
- protease inhibitors
- proteins such as α2-macroglobulin that can bind to proteases and inhibit their enzymatic activity. (Chapter 2)
- defensins
- any member of a large family of small antimicrobial peptides 35–40 amino acids long that can penetrate microbial membranes and disrupt their integrity. They are present at epithelial surfaces and in neutrophil granules. (Chapter 2)
- α-defensins
- a subset of the secreted antimicrobial peptides called defensins. (Chapter 2)
- β-defensins
- a subset of the secreted antimicrobial peptides called defensins. (Chapter 2)
- pentraxins
- any member of a family of pentameric proteins that circulate in the blood and lymph and can bind to the surfaces of a variety of pathogens and target them for destruction. C-reactive protein is a pentraxin. (Chapter 2)
- α2-macroglobulin
- a protease inhibitor in plasma that is a component of innate immunity. It inhibits proteases that are produced or acquired by bacteria to aid their invasion. (Chapter 2)