7.8 Summary and Overview of the SN2 and SN1 Reactions
These two substitution reactions are interconnected by more than the ultimate formation of a substitution product. They complement each other—when one is ineffective, the other is likely to work well. For example, the SN2 reaction works best for methyl and primary substrates, which is exactly when the SN1 reaction is least effective. When the SN2 reaction fails, as with tertiary substrates, the SN1 reaction is most effective (Fig. 7.82).
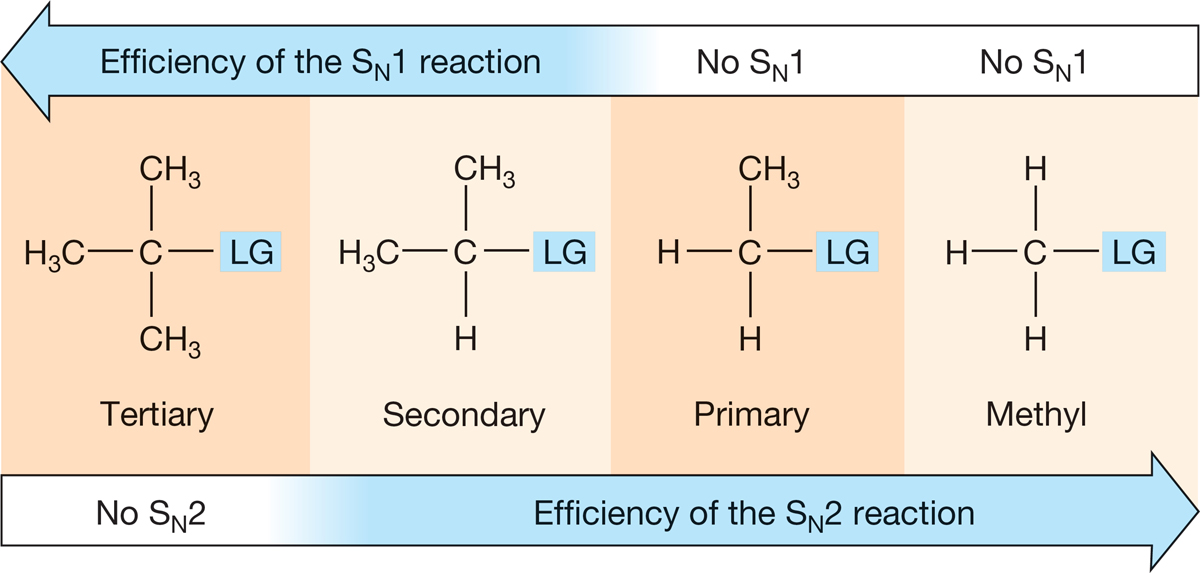
FIGURE 7.82 The SN2 and SN1 reactions are complementary.
At the extremes, the situation is clear. The SN2 reaction dominates for methyl and primary substrates because there is little steric hindrance to the displacement of the leaving group and because the competing SN1 reaction is disfavored by the necessity of forming the highly unstable methyl or primary carbocations (Fig. 7.83).
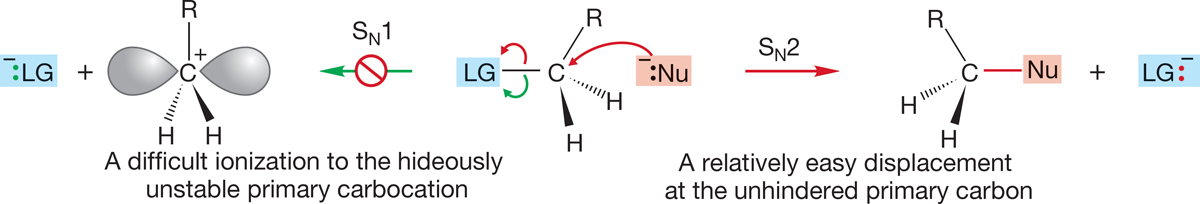
FIGURE 7.83 A comparison of the SN1 and SN2 reactions for primary substrates.
The SN1 reaction dominates for tertiary substrates because ionization can produce the relatively stable tertiary carbocation and because the competing SN2 reaction is strongly disfavored by the steric hindrance to the nucleophile approaching from the rear (Fig. 7.84).
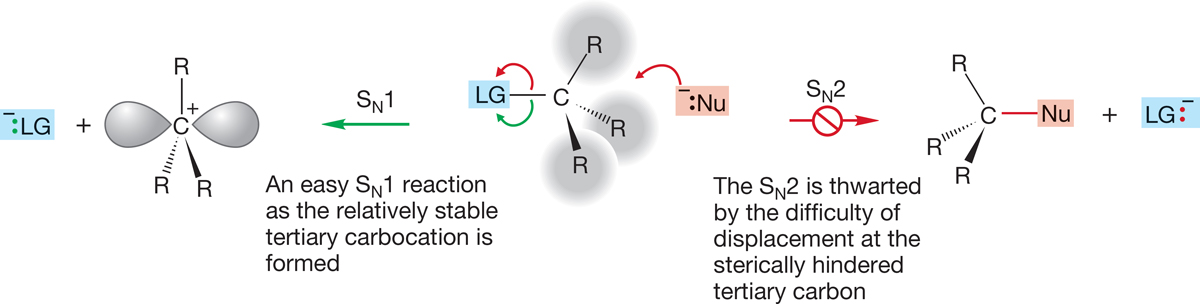
FIGURE 7.84 For tertiary systems, the SN1 reaction dominates: The ionization produces the relatively stable tertiary carbocation, and the SN2 reaction is hindered by the three R groups that thwart approach of the nucleophile from the rear.
In the middle ground—secondary substrates—we might expect to be able to find both reactions. Small R groups, use of a powerful nucleophile, and a solvent of proper polarity for the charge type of the reaction favor the SN2 reaction. Large R groups, a weak nucleophile, and a polar solvent favor the SN1 reaction (Fig. 7.85).
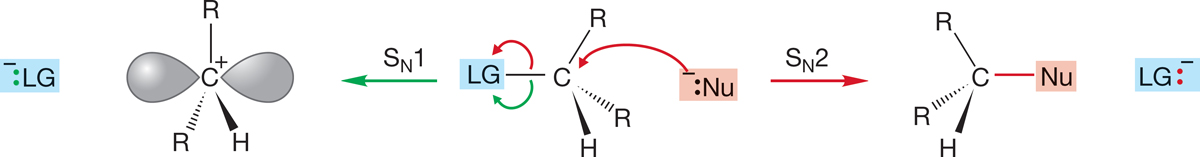
FIGURE 7.85 For secondary substrates, both SN1 and SN2 reactions may be possible.
PROBLEM 7.26 Add appropriate reagents and solvents for each transformation:
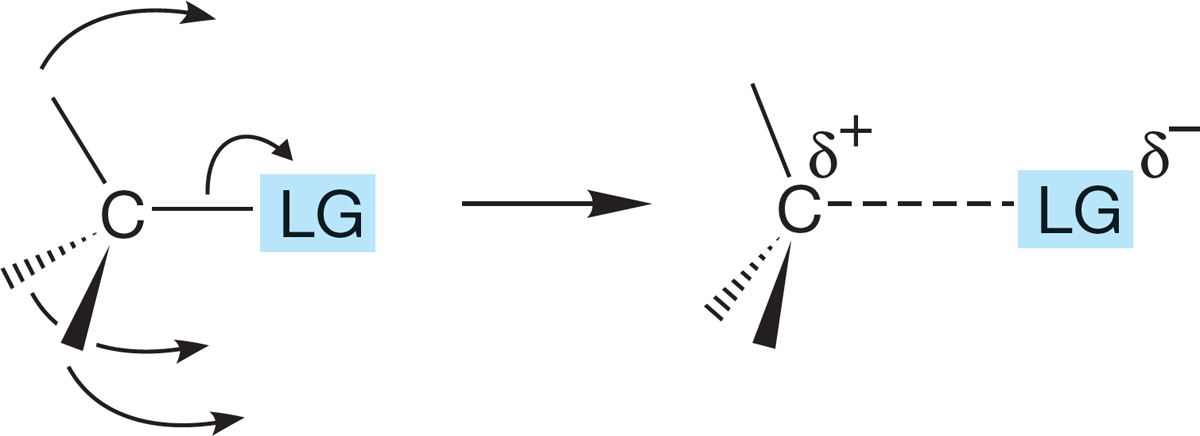
FIGURE 7.86 The beginning of an SN1 reaction. The bond to the leaving group :LG− has lengthened, and the tetrahedron has begun to flatten out. The hybridization of the central carbon is changing from nearly sp3 to the sp2 of the planar carbocation.
We have given a picture of these reactions that emphasizes extremes, developing an “either SN2 or SN1” view. However, the situation is messier than this simple explanation would suggest, especially for secondary compounds. Imagine beginning to ionize a bond to a leaving group. As the bond stretches, positive charge begins to develop on carbon (δ+), and rehybridization toward sp2 begins as the groups attached to the carbon flatten out (Fig. 7.86).
If we simply continue the process of ionization, we have the pure SN1 reaction. If we let a nucleophilic solvent molecule interact with the rear of the departing bond and ultimately form a bond to carbon, we have the SN2 reaction with inversion (Fig. 7.87).
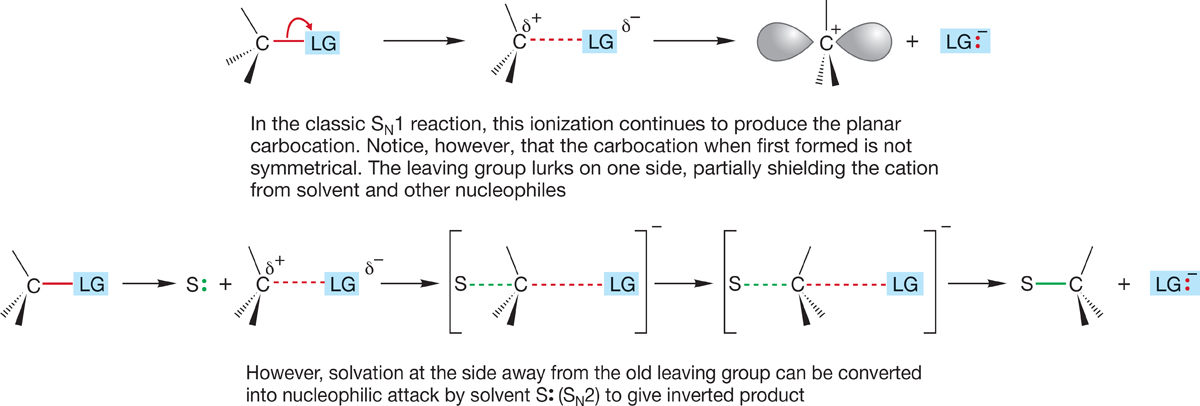
FIGURE 7.87 Especially for secondary substrates, the difference between SN2, displacement by solvent, and preferential solvation at the rear of the departing bond is a subtle one.
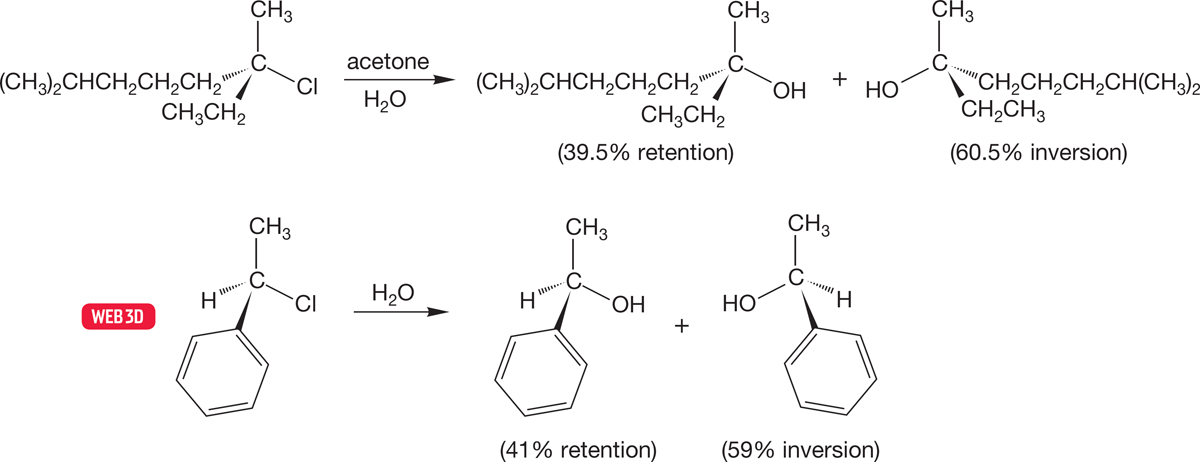
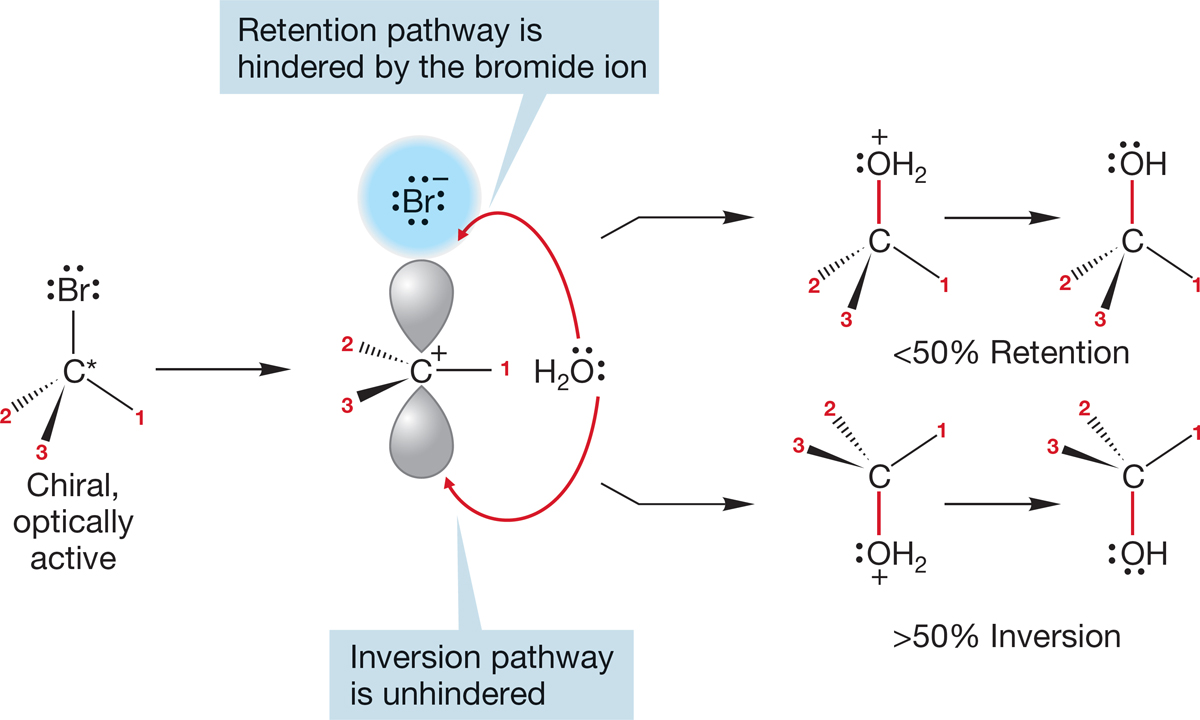
How strong an interaction with the rear lobe of the orbital is needed before we call it bonding? In polar solvents, ions are highly solvated, and in an SN1 reaction solvation will begin to be important as soon as a δ+ charge begins to build up on carbon. Solvation will be especially easy at the rear of the departing group as the leaving group shields the front side. We have already seen that the leaving group can hinder reaction at one side of the developing cation and produce some net inversion in the reaction (Figs. 7.76 and 7.77).
Now we begin to see the difficulty of telling the two mechanisms apart in some situations. When does “preferential solvation from the rear” become “displacement from the rear”? It becomes painfully hard to sort out in some cases. Organic chemistry is frustratingly, if delightfully, messy.
This messiness raises its head in other ways. Almost all substitution reactions are accompanied by some formation of alkene. In organic chemistry, few reactions proceed in 100% yield, quantitatively producing one molecule from another. A large part of the business of organic synthesis is the search for specificity. Synthetic chemists seek reagents and reaction conditions that greatly favor one reaction pathway over others. In particular, attempts to synthesize molecules through substitution reactions must take account of competitive reactions called eliminations. A hydrogen atom and a leaving group are lost (eliminated) from the substrate to give an alkene (Fig. 7.88). In the next chapter, we examine two general mechanisms for these elimination reactions.
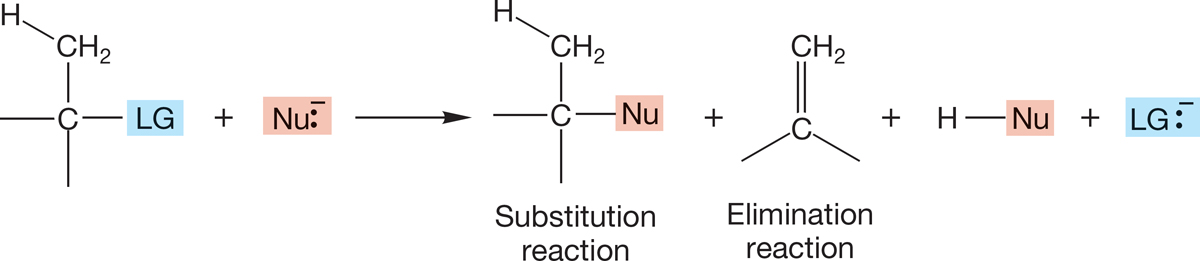
FIGURE 7.88 In almost all SN1 and SN2 reactions, substitution is accompanied by some elimination of H―LG to give an alkene.