HOW PROTEINS ARE STUDIED
Understanding how a particular protein functions calls for detailed structural and biochemical analyses—both of which require large amounts of pure protein. But isolating a single type of protein from the thousands of other proteins present in a cell is a formidable task. For many years, proteins had to be purified directly from the source—the tissues in which they are most plentiful. That approach was inconvenient, entailing, for example, early-morning trips to the slaughterhouse. More importantly, the complexity of intact tissues and organs is a major disadvantage when trying to purify particular molecules, because a long series of chromatography steps is generally required. These procedures not only take weeks to perform, but they also yield only a few milligrams of pure protein.
Nowadays, proteins are more often isolated from cells that are grown in a laboratory (see, for example, Figure 1−39). Often these cells have been “tricked” into making large quantities of a given protein using the genetic engineering techniques discussed in Chapter 10. Such engineered cells frequently allow large amounts of pure protein to be obtained in only a few days.
In this section, we outline how proteins are extracted and purified from cultured cells and other sources. We describe how these proteins are analyzed to determine their amino acid sequence and their three-dimensional structure. Finally, we discuss how technical advances are allowing proteins to be analyzed, cataloged, manipulated, and even designed from scratch.
Proteins Can Be Purified from Cells or Tissues
Whether starting with a piece of liver or a vat of bacteria, yeast, or animal cells that have been engineered to produce a protein of interest, the first step in any purification procedure involves breaking open the cells to release their contents. The resulting slurry is called a cell homogenate or extract. This physical disruption is followed by an initial fractionation procedure to separate out the class of molecules of interest—for example, all the soluble proteins in the cell (Panel 4−3, pp. 164–165).
With this collection of proteins in hand, the job is then to isolate the desired protein. The standard approach involves purifying the protein through a series of chromatography steps, which use different materials to separate the individual components of a complex mixture into portions, or fractions, based on the properties of the protein—such as size, shape, or electrical charge. After each separation step, the resulting fractions are examined to determine which ones contain the protein of interest. These fractions are then pooled and subjected to additional chromatography steps until the desired protein is obtained in pure form.
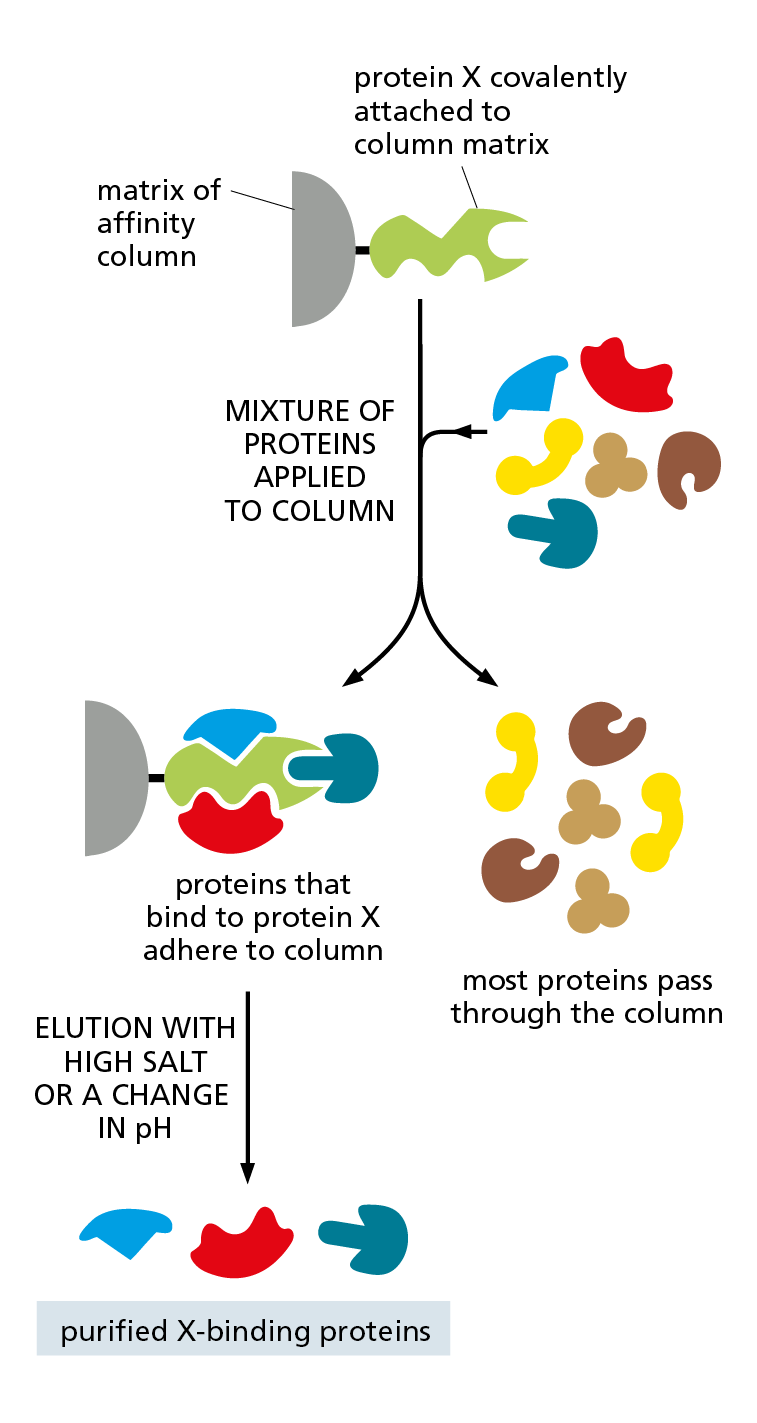
Figure 4−55 Affinity chromatography can be used to isolate the binding partners of a protein of interest. The purified protein of interest (protein X) is covalently attached to the matrix of a chromatography column. An extract containing a mixture of proteins is then loaded onto the column. Those proteins that associate with protein X inside the cell will usually bind to it on the column. Proteins not bound to the column pass right through, and the proteins that are bound tightly to protein X can then be released by changing the pH or ionic composition of the washing solution.
The most efficient forms of protein chromatography separate polypeptides on the basis of their ability to bind to a particular molecule—a process called affinity chromatography (Panel 4−4, p. 166). If large amounts of antibodies that recognize the protein are available, for example, they can be attached to the matrix of a chromatography column and used to help extract the protein from a mixture (see Panel 4−2, pp. 140–141).
Affinity chromatography can also be used to isolate proteins that interact physically with a protein being studied. In this case, the purified protein of interest is attached tightly to the column matrix; the proteins that bind to it will remain in the column and can then be removed by changing the composition of the washing solution (Figure 4−55).
Proteins can also be separated by electrophoresis. In this technique, a mixture of proteins is loaded onto a polymer gel and subjected to an electric field; the polypeptides will then migrate through the gel at different speeds depending on their size and net charge (Panel 4−5, p. 167). If too many proteins are present in the sample, or if the proteins are very similar in their migration rate, they can be resolved further using two-dimensional gel electrophoresis (see Panel 4−5). These electrophoretic approaches yield a number of bands or spots that can be visualized by staining; each band or spot contains a different protein. Chromatography and electrophoresis—both developed more than 70 years ago but greatly improved since—continue to be instrumental in building an understanding of what proteins look like and how they behave. These and other historical breakthroughs are described in Table 4−2.
|
Table 4–2 historical landmarks in our understanding of proteins |
|
|
1838 |
The name “protein” (from the Greek proteios, “primary”) was suggested by Berzelius for the complex nitrogen-rich substance found in the cells of all animals and plants |
|
1819–1904 |
Most of the 20 common amino acids found in proteins were discovered |
|
1864 |
Hoppe-Seyler crystallized, and named, the protein hemoglobin |
|
1894 |
Fischer proposed a lock-and-key analogy for enzyme–substrate interactions |
|
1897 |
Buchner and Buchner showed that cell-free extracts of yeast can break down sucrose to form carbon dioxide and ethanol, thereby laying the foundations of enzymology |
|
1926 |
Sumner crystallized urease in pure form, demonstrating that proteins could possess the catalytic activity of enzymes; Svedberg developed the first analytical ultracentrifuge and used it to estimate the correct molecular weight of hemoglobin |
|
1933 |
Tiselius introduced electrophoresis for separating proteins in solution |
|
1934 |
Bernal and Crowfoot presented the first detailed x-ray diffraction patterns of a protein, obtained from crystals of the enzyme pepsin |
|
1942 |
Martin and Synge developed chromatography, a technique now widely used to separate proteins |
|
1951 |
Pauling and Corey proposed the structure of a helical conformation of a chain of amino acids—the α helix—and the structure of the β sheet, both of which were later found in many proteins |
|
1955 |
Sanger determined the order of amino acids in insulin, the first protein whose amino acid sequence was determined |
|
1956 |
Ingram produced the first protein fingerprints, showing that the difference between sickle-cell hemoglobin and normal hemoglobin is due to a change in a single amino acid (Movie 4.13) |
|
1960 |
Kendrew described the first detailed three-dimensional structure of a protein (sperm whale myoglobin) to a resolution of 0.2 nm, and Perutz proposed a lower-resolution structure for hemoglobin |
|
1963 |
Monod, Jacob, and Changeux recognized that many enzymes are regulated through allosteric changes in their conformation |
|
1966 |
Phillips described the three-dimensional structure of lysozyme by x-ray crystallography, the first enzyme to be analyzed in atomic detail |
|
1973 |
Nomura reconstituted a functional bacterial ribosome from purified components |
|
1975 |
Henderson and Unwin determined the first three-dimensional structure of a transmembrane protein (bacteriorhodopsin), using a computer-based reconstruction from electron micrographs |
|
1976 |
Neher and Sakmann developed patch-clamp recording to measure the activity of single ion-channel proteins |
|
1984 |
Wüthrich used nuclear magnetic resonance (NMR) spectroscopy to solve the three-dimensional structure of a soluble sperm protein |
|
1988 |
Tanaka and Fenn separately developed methods for using mass spectrometry to analyze proteins and other biological macromolecules |
|
1996–2013 |
Mann, Aebersold, Yates, and others refine methods for using mass spectrometry to identify proteins in complex mixtures, exploiting the availability of complete genome sequences |
|
1975–2013 |
Frank, Dubochet, Henderson and others develop computer-based methods for single-particle cryoelectron microscopy (cryo-EM), enabling determination of the structures of large protein complexes at atomic resolution |
Once a protein has been obtained in pure form, it can be used in biochemical assays to study the details of its activity. It can also be subjected to techniques that reveal its amino acid sequence and, ultimately, its precise three-dimensional structure.
Determining a Protein’s Structure Begins with Determining Its Amino Acid Sequence
The task of determining a protein’s primary structure—its amino acid sequence—can be accomplished in several ways. For many years, sequencing a protein was done by directly analyzing the amino acids in the purified protein. First, the protein was broken down into smaller pieces using a selective protease; the enzyme trypsin, for example, cleaves polypeptide chains on the carboxyl side of a lysine or an arginine. Then the identities of the amino acids in each fragment were determined chemically. The first protein sequenced in this way was the hormone insulin in 1955.
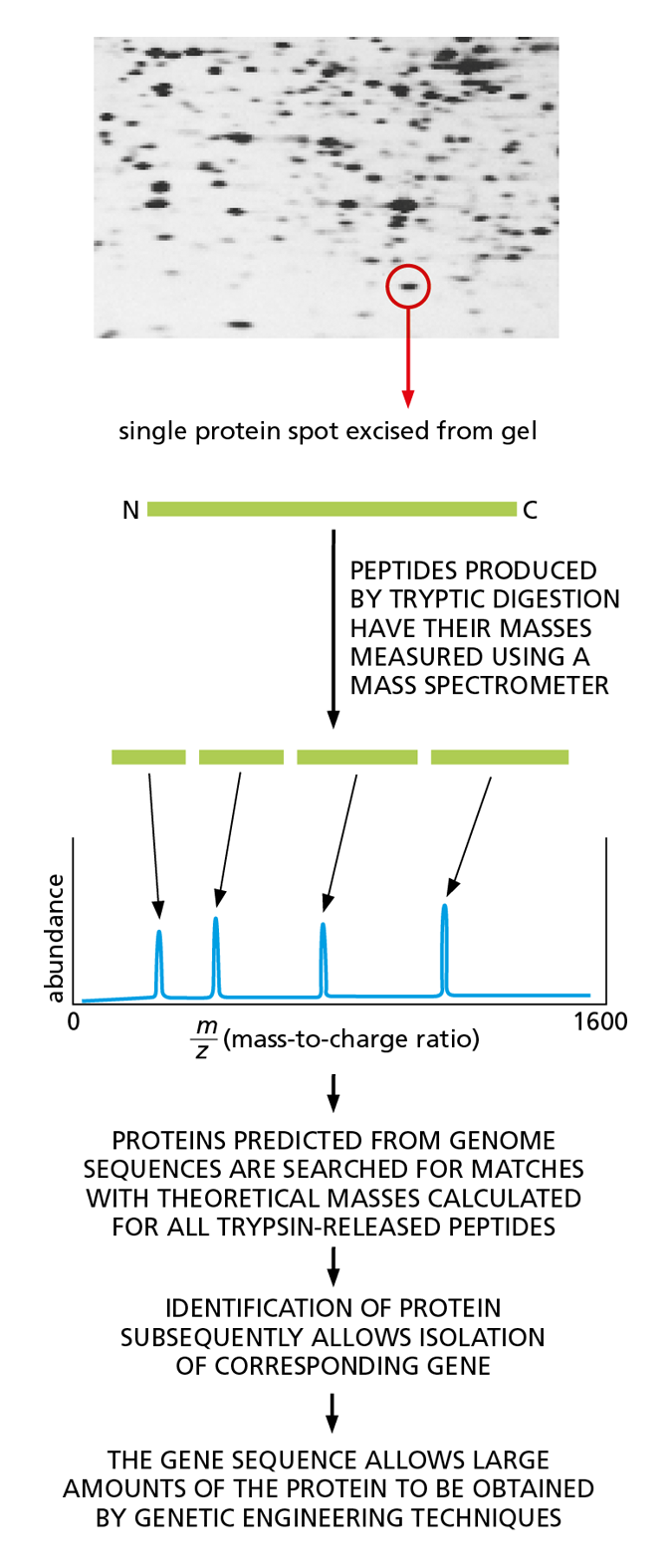
Figure 4−56 Mass spectrometry can be used to identify proteins by determining the precise masses of peptides derived from them. As indicated, this in turn allows proteins of interest to be produced in the large amounts needed for determining their three-dimensional structure. In this example, a protein of interest is excised from a polyacrylamide gel after two-dimensional electrophoresis (see Panel 4−5, p. 167) and then digested with trypsin. The peptide fragments are loaded into the mass spectrometer, and their exact masses are measured. Genome sequence databases are then searched to find the protein encoded by the organism in question whose profile matches this peptide fingerprint. Mixtures of proteins can also be analyzed in this way. (Image courtesy of Patrick O’Farrell.)
A much faster way to determine the amino acid sequence of proteins that have been isolated from organisms for which the full genome sequence is known is a method called mass spectrometry. This technique determines the exact mass of every peptide fragment in a purified protein, which then allows the protein to be identified from a database that contains a list of every protein thought to be encoded by the genome of the relevant organism. Such lists are computed by taking the organism’s genome sequence and applying the genetic code (discussed in Chapter 7).
To perform mass spectrometry, the peptides derived from digestion with trypsin are blasted with a laser. This treatment heats the peptides, causing them to become electrically charged (ionized) and ejected in the form of a gas. Accelerated by a powerful electric field, the peptide ions then fly toward a detector; the time it takes them to arrive is related to their mass and their charge. (The larger the peptide is, the more slowly it moves; the more highly charged it is, the faster it moves.) The set of very exact masses of the protein fragments produced by trypsin cleavage then serves as a “fingerprint” that can be used to identify the protein—and its corresponding gene—from publicly accessible databases (Figure 4−56).
This approach can even be applied to complex mixtures of proteins; for example, starting with an extract containing all the proteins made by yeast cells grown under a particular set of conditions. To obtain the increased resolution required to distinguish individual proteins, such mixtures are frequently analyzed using tandem mass spectrometry. In this case, after the peptides pass through the first mass spectrometer, they are broken into even smaller fragments and analyzed by a second mass spectrometer.
Although all the information required for a polypeptide chain to fold is contained in its amino acid sequence, only in special cases can we reliably predict a protein’s detailed three-dimensional conformation—the spatial arrangement of its atoms—from its sequence alone. Today, the predominant way to discover the precise folding pattern of any protein is by experiment, using x-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, or most recently cryoelectron microscopy (cryo-EM), as described in Panel 4–6 (pp. 168–169).
Genetic Engineering Techniques Permit the Large-Scale Production, Design, and Analysis of Almost Any Protein
Advances in genetic engineering techniques now permit the production of large quantities of almost any desired protein. In addition to making life much easier for biochemists interested in purifying specific proteins, this ability to churn out huge quantities of a protein has given rise to an entire biotechnology industry (Figure 4−57). Bacteria, yeast, and cultured mammalian cells are now used to mass-produce a variety of therapeutic proteins, such as insulin, human growth hormone, and even the fertility-enhancing drugs used to boost egg production in women undergoing in vitro fertilization treatment. Preparing these proteins previously required the collection and processing of vast amounts of tissue and other biological products—including, in the case of the fertility drugs, the urine of postmenopausal nuns.

Figure 4−57 Biotechnology companies produce mass quantities of useful proteins. Shown in this photograph are the large, turnkey microbial fermenters used to produce a whooping cough vaccine. (Courtesy of Pierre Guerin Technologies.)
The same sorts of genetic engineering techniques can also be employed to produce new proteins and enzymes that contain novel structures or perform unusual tasks: metabolizing toxic wastes or synthesizing life-saving drugs, for example. Most synthetic catalysts are nowhere near as effective as naturally occurring enzymes in terms of their ability to speed the rate of selected chemical reactions. But, as we continue to learn more about how proteins and enzymes exploit their unique conformations to carry out their biological functions, our ability to make novel proteins with useful functions can only improve.
The Relatedness of Proteins Aids the Prediction of Protein Structure and Function
Biochemists have made enormous progress over the past 150 years in understanding the structure and function of proteins (see Table 4−2, p. 160). These advances are the fruits of decades of painstaking research on isolated proteins, performed by individual scientists working tirelessly on single proteins or protein families, one by one, sometimes for their entire careers. In the future, however, more and more of these investigations of protein conformation and activity will likely take place on a larger scale.
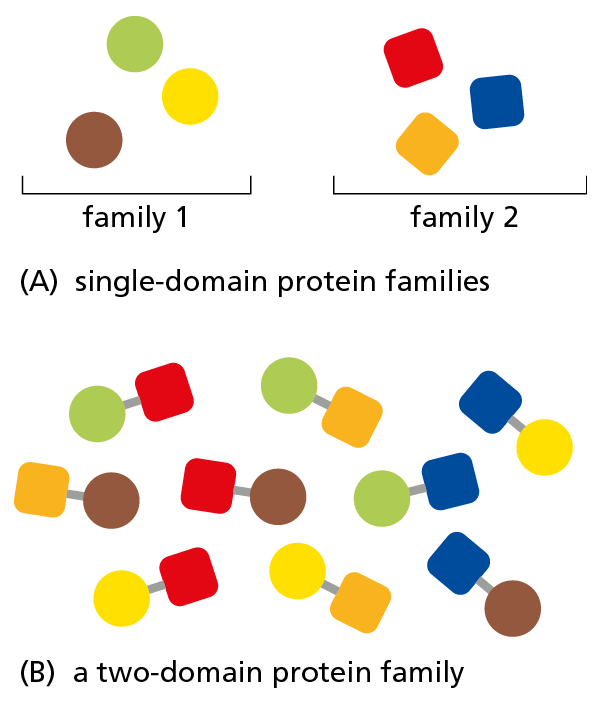
Figure 4−58 Most proteins belong to structurally related families. (A) More than two-thirds of all well-studied proteins contain a single structural domain. The members of these single-domain families can have different amino acid sequences but fold into a protein with a similar shape. (B) During evolution, structural domains have been combined in different ways to produce families of multidomain proteins. Almost all novelty in protein structure comes from the way these single domains are arranged. Unlike the number of novel single domains, the number of multidomain families being added to the public databases is still rapidly increasing.
Improvements in our ability to rapidly sequence whole genomes, and the development of methods such as mass spectrometry, have fueled our ability to determine the amino acid sequences of enormous numbers of proteins. Millions of unique protein sequences from thousands of different species have thereby been deposited into publicly available databases, and the collection is expected to double in size every two years. Comparing the amino acid sequences of all of these proteins reveals that the majority belong to protein families that share specific “sequence patterns”—stretches of amino acids that fold into distinct structural domains. In some of these families, the proteins contain only a single structural domain. In others, the proteins include multiple domains arranged in novel combinations (Figure 4−58).
Although the number of multidomain families is growing rapidly, the discovery of novel single domains appears to be leveling off. This plateau suggests that the vast majority of proteins may fold up into a limited number of structural domains—perhaps as few as 10,000 to 20,000. For many single-domain families, the structure of at least one family member is known. And knowing the structure of one family member allows us to say something about the structure of its relatives. By this account, we have some structural information for almost three-quarters of the proteins archived in databases (Movie 4.14).
A future goal is to acquire the ability to look at a protein’s amino acid sequence and be able to deduce its structure and gain insight into its function. We are coming closer to being able to predict protein structure based on sequence information alone, but we still have a considerable way to go. To date, computational methods that take an amino acid sequence and search for the protein conformations with the lowest energy have been successful for proteins less than 100 amino acids long, or for longer proteins for which additional information is available (such as homology with proteins whose structure is known).
Looking at an amino acid sequence and predicting how a protein will function—alone or as part of a complex in the cell—is more challenging still. But the closer we get to accomplishing these goals, the closer we will be to understanding the fundamental basis of life.
- chromatography
Technique used to separate the individual molecules in a complex mixture on the basis of their size, charge, or their ability to bind to a particular chemical group. In a common form of the technique, the mixture is run through a column filled with a material that binds the desired molecule, and it is then eluted from the column with a solvent gradient.
- electrophoresis
Technique for separating a mixture of proteins or DNA fragments by placing them on a polymer gel and subjecting them to an electric field. The molecules migrate through the gel at different speeds depending on their size and net charge.
- mass spectrometry
Sensitive technique that enables the determination of the exact mass of all of the molecules in a complex mixture.
- x-ray crystallography
Technique used to determine the three-dimensional structure of a protein molecule by analyzing the diffraction pattern produced when a beam of x-rays is passed through an ordered array of the protein.
- nuclear magnetic resonance (NMR) spectroscopy
Technique used for determining the three-dimensional structure of a protein in solution.
- cryoelectron microscopy (cryo-EM)
Technique for observing the detailed structure of a macromolecule at very low temperatures after freezing native structures in ice.