From a chemical point of view, proteins are by far the most structurally complex and functionally sophisticated molecules known. This is perhaps not surprising, once we realize that the structure and chemistry of each protein have been developed and fine-tuned over billions of years of evolutionary history. The theoretical calculations of population geneticists reveal that, over evolutionary time periods, a surprisingly small selective advantage is enough to cause a randomly altered protein sequence to spread through a population of organisms. Yet, even to experts, the remarkable versatility of proteins can seem truly amazing.
In this section, we consider how the location of each amino acid in a protein’s long string of amino acids determines its three-dimensional shape. Later in the chapter, we use this understanding of protein structure at the atomic level to describe how the precise shape of each protein molecule determines its function in a cell.
The Structure of a Protein Is Specified by Its Amino Acid Sequence
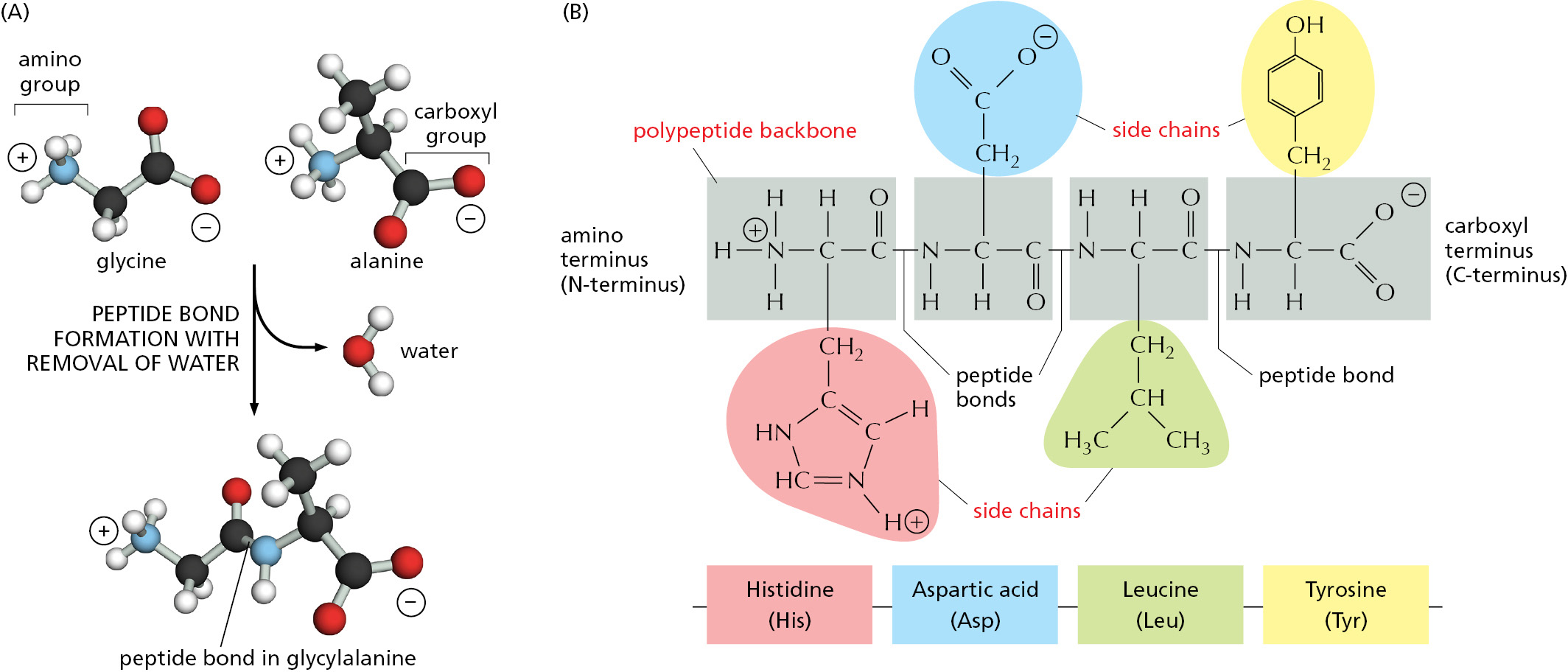
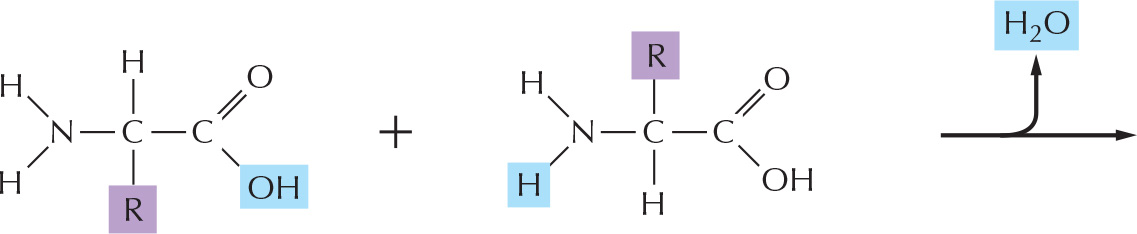
There are 20 different types of amino acids in proteins that are encoded directly in an organism’s DNA, each with different chemical properties. Every protein molecule consists of a long unbranched chain of these amino acids, each linked to its neighbor through a covalent peptide bond (Figure 3–1A). Proteins are therefore also known as polypeptides. Each type of protein has a unique sequence of amino acids, and there are many thousands of different proteins in a cell.
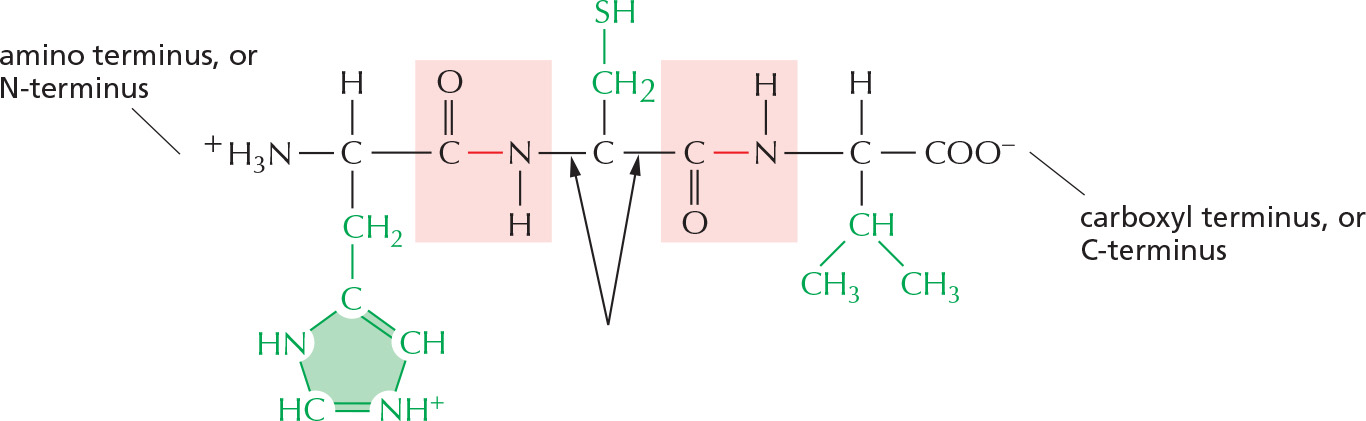
Figure 3–1The components of a protein. (A) Formation of a peptide bond. This covalent bond forms when the carbon atom of the carboxyl group of one amino acid (such as glycine) shares electrons with the nitrogen atom from the amino group of a second amino acid (such as alanine). As indicated, a molecule of water is eliminated in this condensation reaction (see Figure 2–9). In this model, carbon atoms are black, nitrogen blue, oxygen red, and hydrogen white. (B) A two-dimensional representation of a short section of polypeptide backbone with its attached side chains. Each type of protein differs in its sequence and number of amino acids; it is the sequence of the chemically different side chains that makes each protein distinct. The two ends of a polypeptide chain are chemically different: the end carrying the free amino group (NH2, which takes up a proton at neutral pH to become NH3+) is the amino terminus, or N-terminus, and the end carrying the free carboxyl group (COOH, which loses a proton at neutral pH to become COO–) is the carboxyl terminus, or C-terminus. Note that, for simplicity, in many figures in this textbook, NH2 and COOH are used to denote these termini, instead of their actual ionized forms. The amino acid sequence of a protein is always presented in the N-to-C direction, reading from left to right.
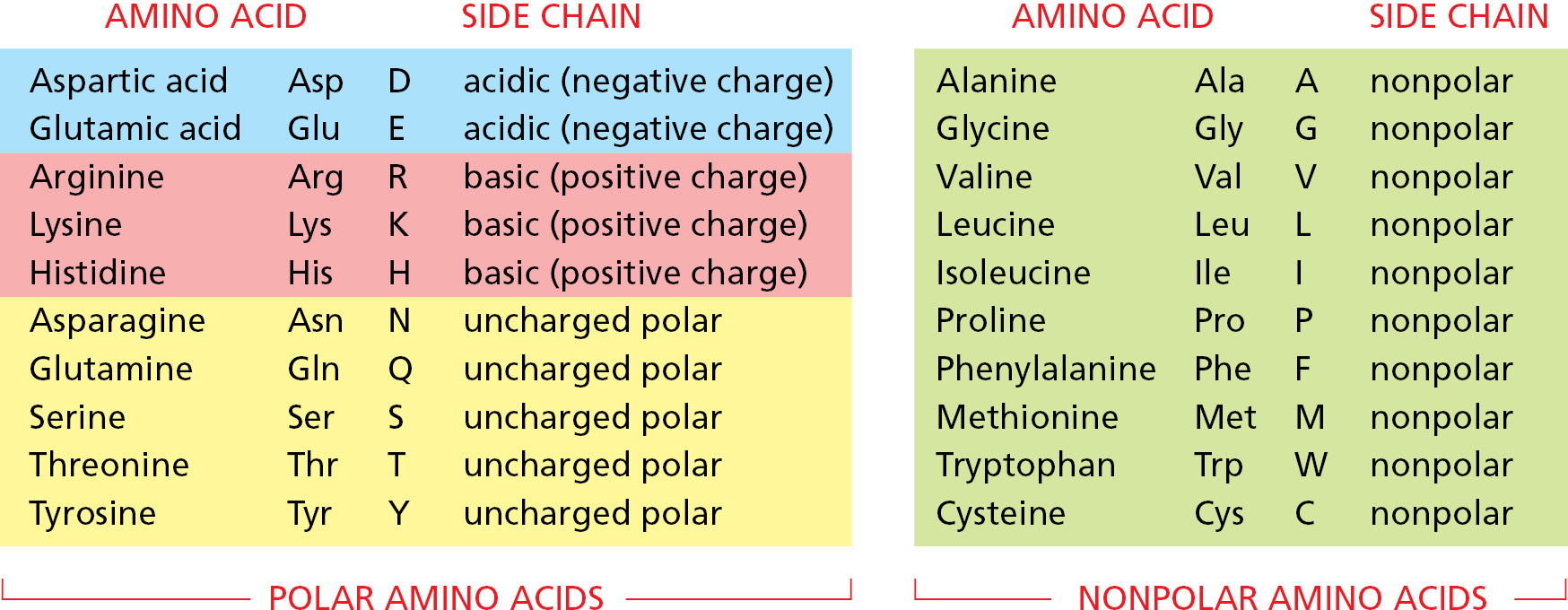
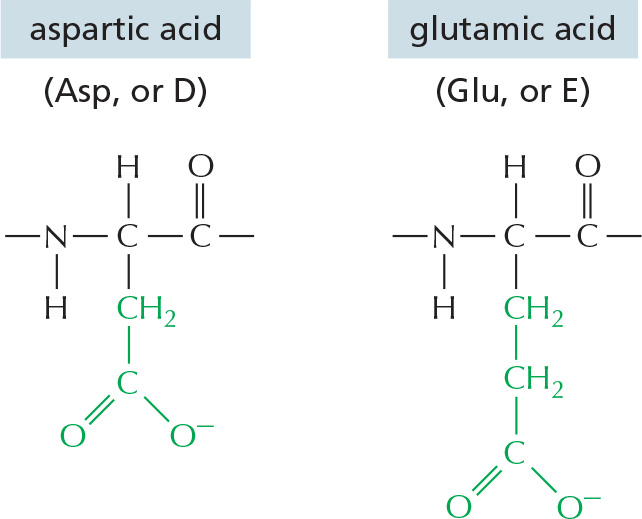
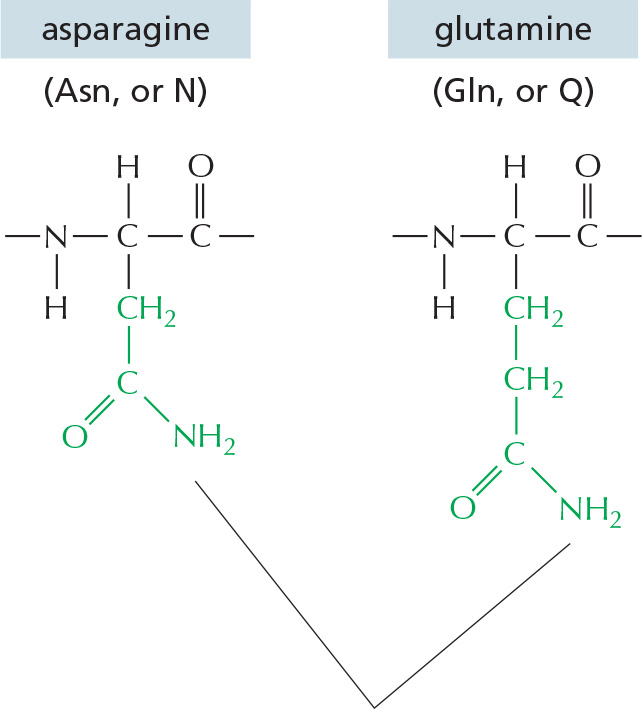
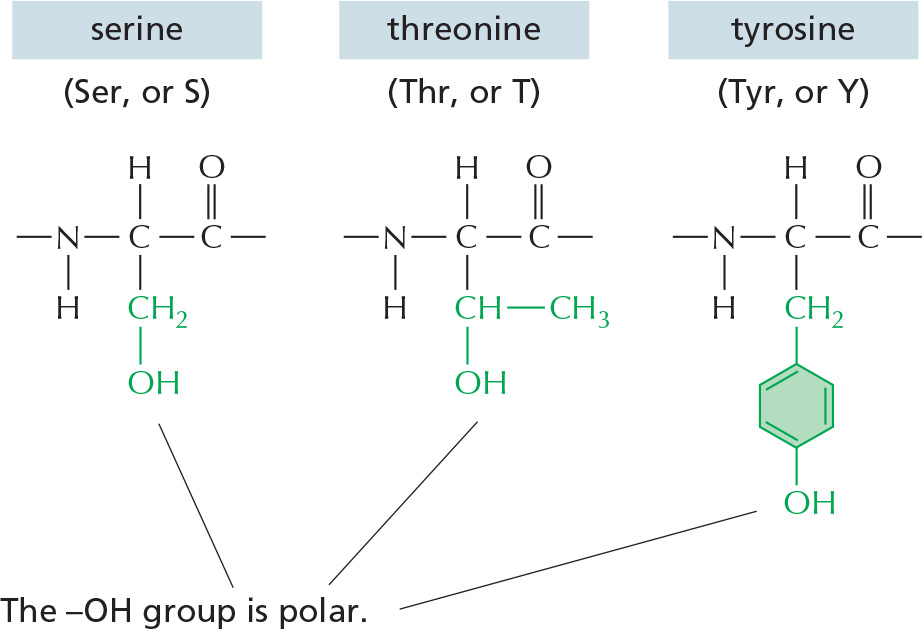
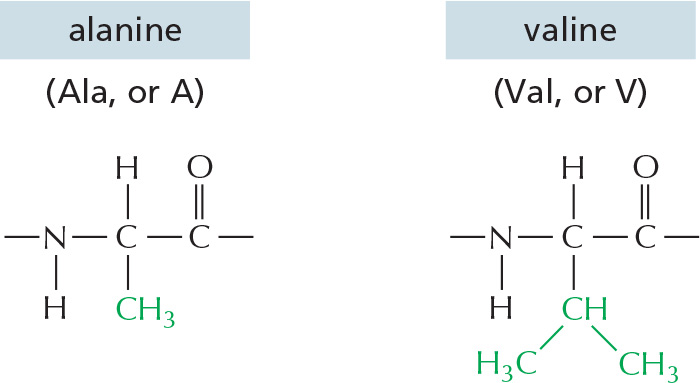
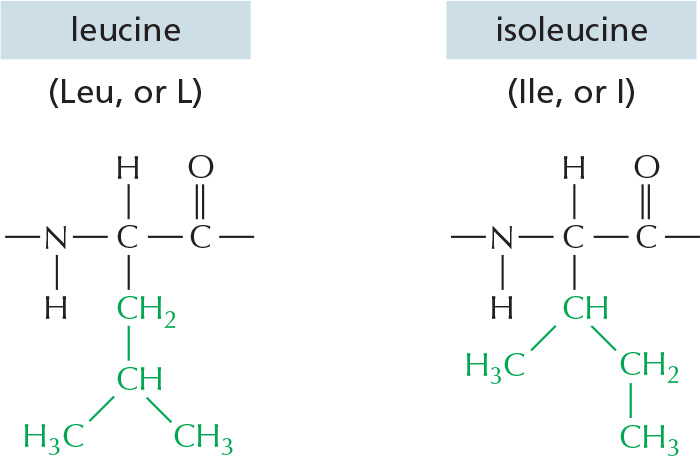
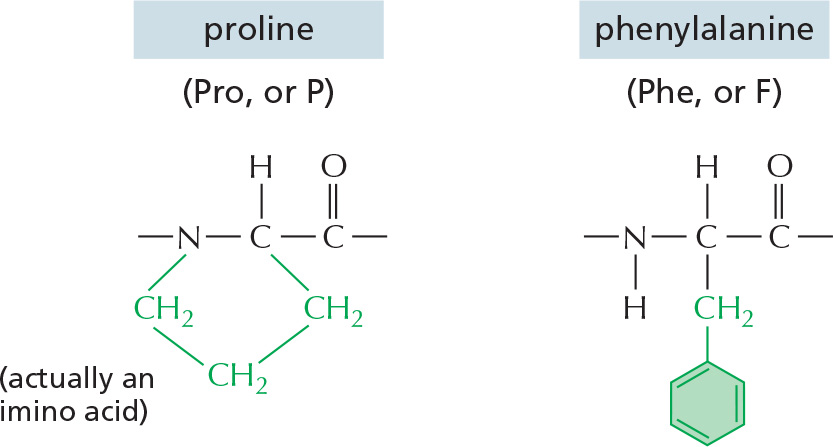
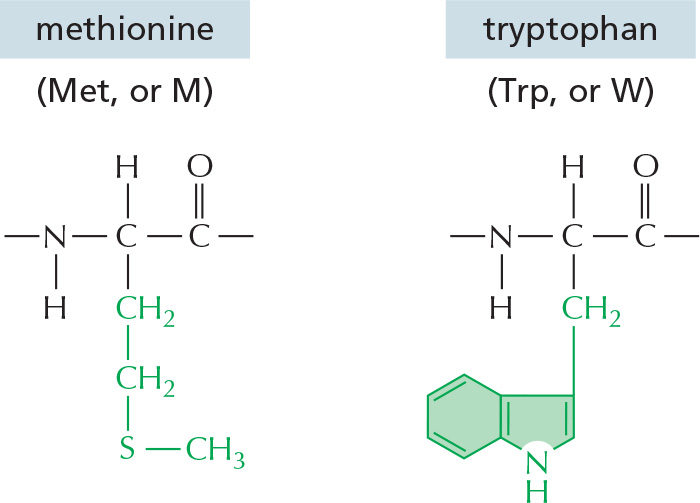
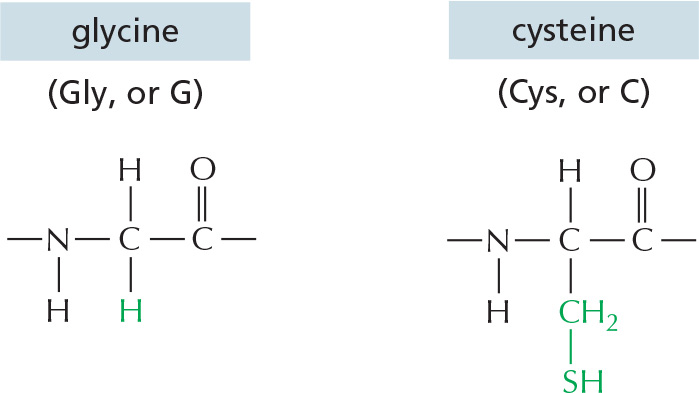
The repeating sequence of atoms along the core of the polypeptide chain is referred to as the polypeptide backbone. Attached to this repetitive backbone are those portions of the amino acids that are not involved in making a peptide bond; these are the 20 different amino acid side chains that give each amino acid its unique properties (Figure 3–1B). Some of these side chains are nonpolar and hydrophobic (“water-fearing”), others are negatively or positively charged, some can readily form covalent bonds, and so on. Panel 3–1 (pp. 118–119) shows their atomic structures, and Figure 3–2 lists their abbreviations.
Figure 3–2The 20 amino acids commonly found in proteins. Each amino acid has a three-letter and a one-letter abbreviation. There are equal numbers of polar and nonpolar side chains; however, some side chains listed here as polar are large enough to have some nonpolar properties (for example, Thr, Tyr, Arg, Lys). For atomic structures, see Panel 3–1 (pp. 118–119).
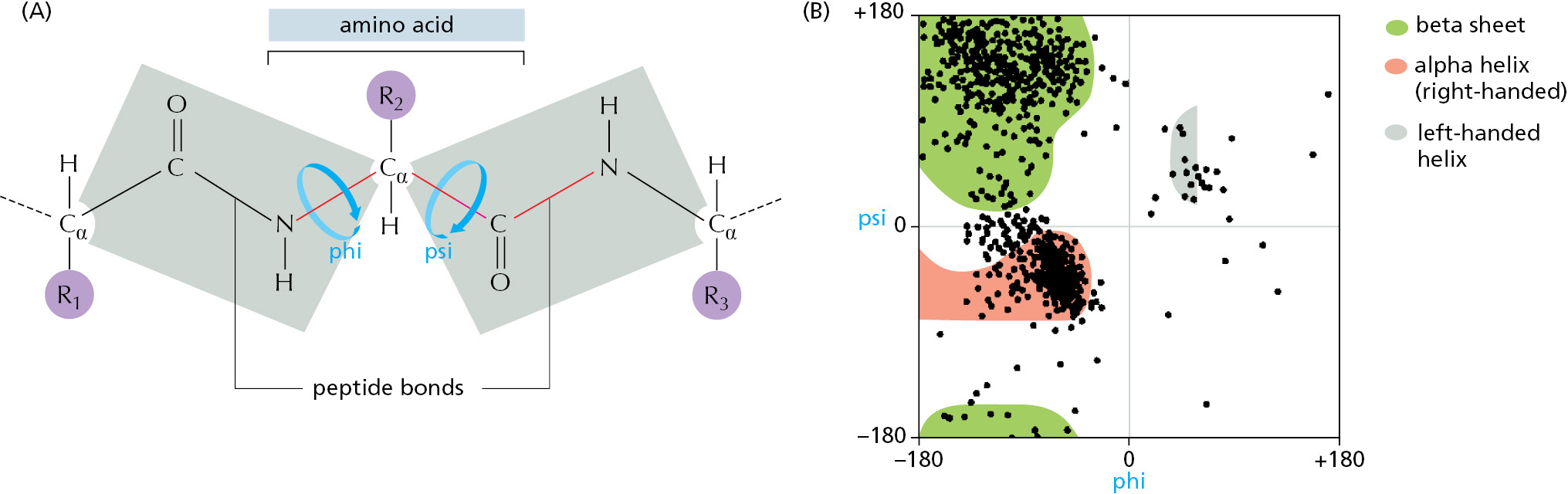
As discussed in Chapter 2, atoms behave almost as if they were hard spheres with a definite radius (their van der Waals radius). Other constraints limit the possible bond angles in a polypeptide chain, and this—plus the requirement that no two atoms overlap—severely restricts the possible three-dimensional arrangements (or conformations) of proteins. As illustrated in Figure 3–3, these steric restrictions (which include a delocalization of electrons in the peptide bond that makes that linkage planar) confine the energy minima for the bond angles in polypeptides to a narrow range. But a long flexible chain such as a protein can still fold in an enormous number of different ways.
Figure 3–3Steric limitations on the bond angles in a polypeptide chain. (A) Each amino acid contributes three bonds (red) to the backbone of the chain. Because it has a partial double-bond character, the peptide bond is planar (gray shading) and does not permit free rotation. By contrast, rotation can occur about the Cα–C bond, whose angle of rotation is called psi (Ψ), and about the N–Cα bond, whose angle of rotation is called phi (ϕ). By convention, an R group is often used to denote an amino acid side chain (purple circles). (B) The conformation of the main-chain atoms in a protein is determined by one pair of ϕ and Ψ angles for each amino acid; because of steric restrictions, most of the possible pairs of ϕ and Ψ angles do not occur. In this so-called Ramachandran plot, each dot represents an observed pair of angles in a protein. The three differently shaded clusters of dots reflect three different secondary structures repeatedly found in proteins. Most prominent are the alpha helix and the beta sheet, as will be described in the text. (B, from J. Richardson, Adv. Prot. Chem. 34:174–175, 1981. With permission from Elsevier.)
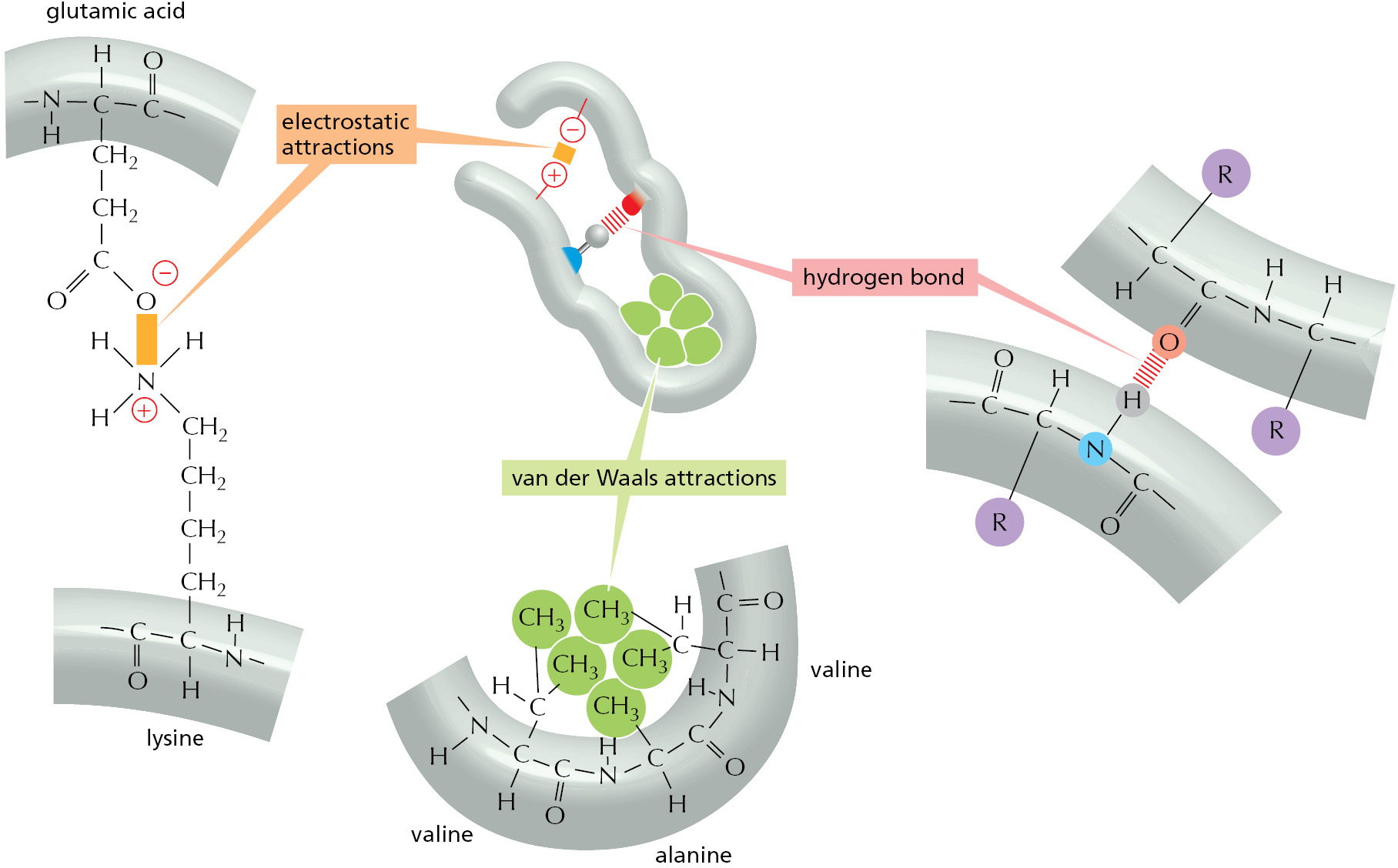
The folding of a protein chain is determined by many different sets of weak noncovalent bonds that form between one part of the chain and another. These involve atoms in the polypeptide backbone, as well as atoms in the amino acid side chains. There are three types of these weak bonds: hydrogen bonds, electrostatic attractions, and van der Waals attractions, as explained in Chapter 2 (see p. 51). Individual noncovalent bonds are 30–300 times weaker than the typical covalent bonds that create biological molecules. But many weak bonds acting in parallel can hold two regions of a polypeptide chain tightly together. It is the combined strength of large numbers of these noncovalent bonds that stabilizes each protein’s folded shape (Figure 3–4).
Figure 3–4Three types of noncovalent bonds help proteins fold. Although a single one of these bonds is quite weak, many of them often act together to create a strong bonding arrangement, as in the example shown. As in the previous figure, R is used as a general designation for an amino acid side chain.
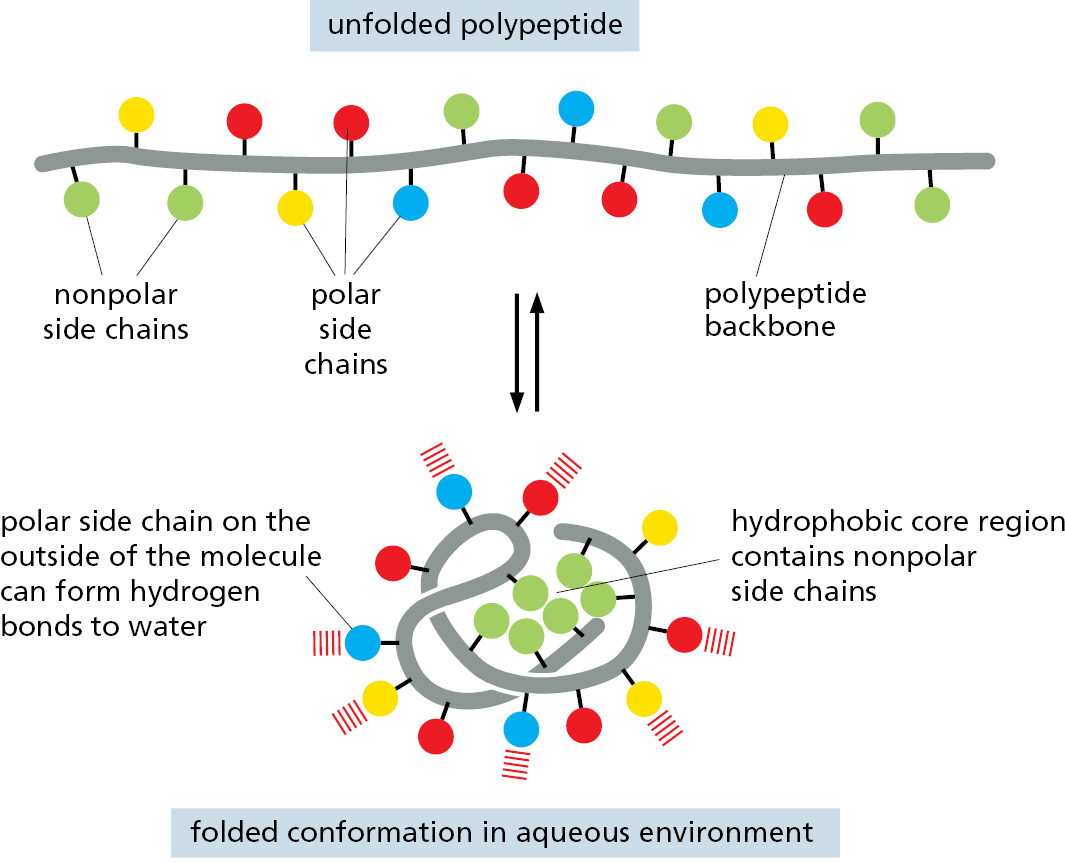
A fourth weak force—a hydrophobic clustering force—also has a central role in determining the shape of a protein. As described in Chapter 2, hydrophobic molecules, including the nonpolar side chains of particular amino acids, tend to be forced together in an aqueous environment in order to minimize their disruptive effect on the hydrogen-bonded network of water molecules (see Panel 2–2, pp. 96–97). Therefore, an important factor governing the folding of any protein is the distribution of its polar and nonpolar amino acids. The nonpolar (hydrophobic) side chains in a protein—belonging to such amino acids as phenylalanine, leucine, valine, and tryptophan—tend to cluster in the interior of the molecule (just as hydrophobic oil droplets coalesce in water to form one large droplet). This enables these side chains to avoid contact with the water that surrounds them inside a cell. In contrast, polar groups—such as those belonging to arginine, glutamine, and histidine—tend to arrange themselves near the outside of the molecule, where they can form hydrogen bonds with water and with other polar molecules (Figure 3–5). Any polar amino acids that are left buried within the protein are usually hydrogen-bonded to other polar amino acids or to the polypeptide backbone.
Figure 3–5How a protein folds into a compact conformation. The polar amino acid side chains tend to lie on the outside of the protein, where they can interact with water; the nonpolar amino acid side chains are buried on the inside forming a tightly packed hydrophobic core of atoms that are hidden from water. In this highly schematic drawing, the protein contains only 17 amino acids; actual proteins are generally much larger.
PANEL 3–1: The 20 Amino Acids Found in Proteins
FAMILIES OF AMINO ACIDS
The common amino acids are grouped according to whether their side chains are
acidicbasicuncharged polarnonpolar
These 20 amino acids are given both three-letter and one-letter abbreviations.
Thus: alanine = Ala = A
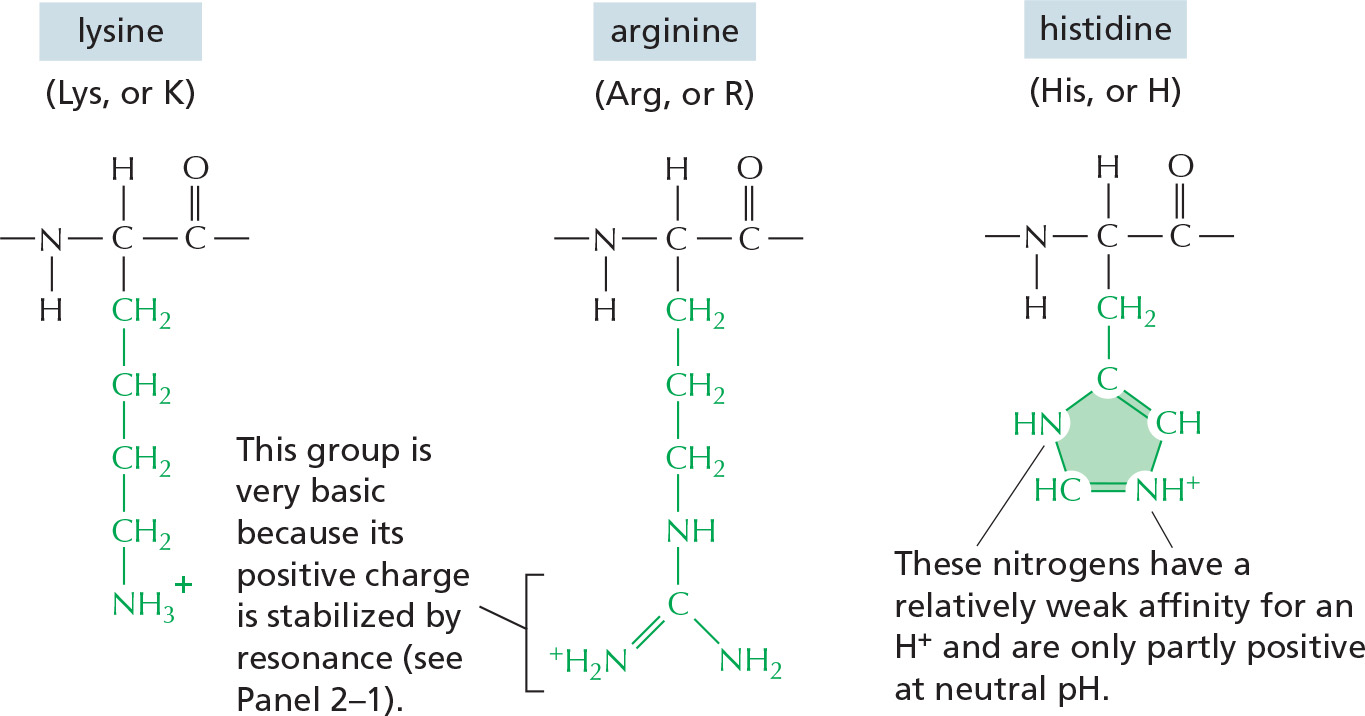
BASIC SIDE CHAINS
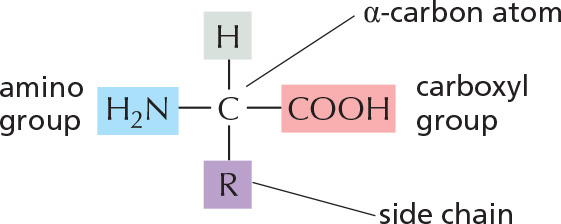
THE AMINO ACID
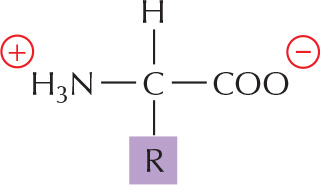
The general formula of an amino acid is
R is commonly one of 20 different side chains. At pH 7, both the amino and carboxyl groups are ionized.
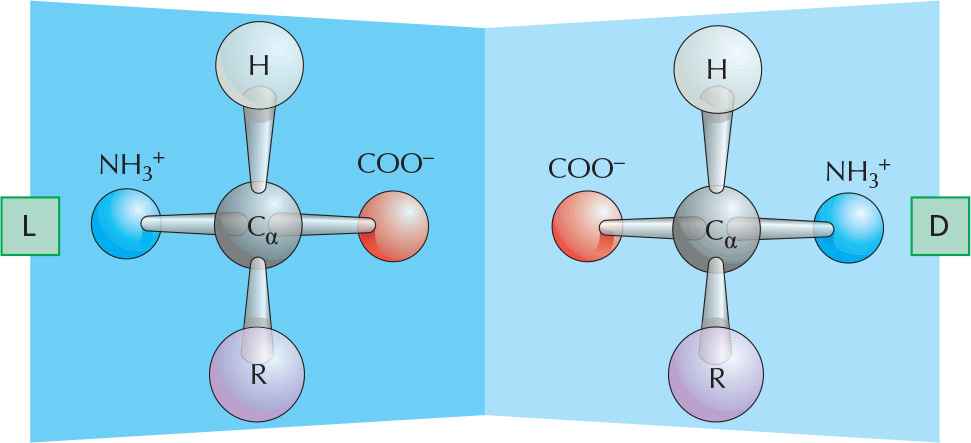
OPTICAL ISOMERS
The α-carbon atom is asymmetric, allowing for two mirror-image (or stereo-) isomers, L and D.
Proteins contain exclusively L-amino acids.
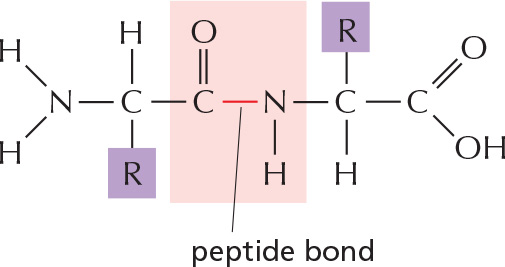
PEPTIDE BONDS
In proteins, amino acids are joined together by an amide linkage, called a peptide bond.
The four atoms involved in each peptide bond form a rigid planar unit (red box). There is no rotation around the C–N bond.
Proteins are long polymers of amino acids linked by peptide bonds, and they are always written with the N-terminus toward the left. Peptides are shorter, usually fewer than 50 amino acids long. The sequence of this tripeptide is histidine-cysteine-valine.
These two single bonds allow rapid rotation, so that long chains of amino acids are very flexible.
ACIDIC SIDE CHAINS
UNCHARGED POLAR SIDE CHAINS
Although the amide N is not charged at neutral pH, it is polar.
NONPOLAR SIDE CHAINS
A disulfide bond(red) can form between two cysteine side chains in proteins.
Proteins Fold into a Conformation of Lowest Energy
As a result of all of these interactions, most proteins have a particular three-dimensional structure, which is determined by the order of the amino acids in a protein’s chain. The final folded structure, or conformation, of any polypeptide chain is generally the one that minimizes its free energy. Biologists have studied protein folding in a test tube using highly purified proteins. Treatment with certain solvents, which disrupt the noncovalent interactions holding the folded chain together, unfolds, or denatures, a protein. This treatment converts the protein into a flexible polypeptide chain that has lost its natural shape. When the denaturing solvent is removed, the protein often refolds spontaneously, or renatures, into its original conformation. This indicates that the amino acid sequence contains all of the information needed for specifying the three-dimensional shape of a protein, a critical point for understanding cell biology.
Most proteins fold up into a single stable conformation. However, this conformation is very dynamic, experiencing constant fluctuations caused by thermal energy. In addition, a protein’s conformation can change when the protein interacts with other molecules in the cell. This change in shape is often crucial to the function of the protein, as we explain in detail later.
Although a protein chain can fold into its correct conformation without outside help, special proteins called molecular chaperones often assist in protein folding (see Chapter 6). Molecular chaperones bind to partly folded polypeptide chains and help them progress along the most energetically favorable folding pathway. In the crowded conditions of the cytoplasm, chaperones are required to prevent the temporarily exposed hydrophobic regions in newly synthesized protein chains from associating with each other to form protein aggregates. However, the final three-dimensional shape of the protein is still specified by its amino acid sequence: chaperones simply make reaching the folded state more reliable.
The α Helix and the β Sheet Are Common Folding Motifs
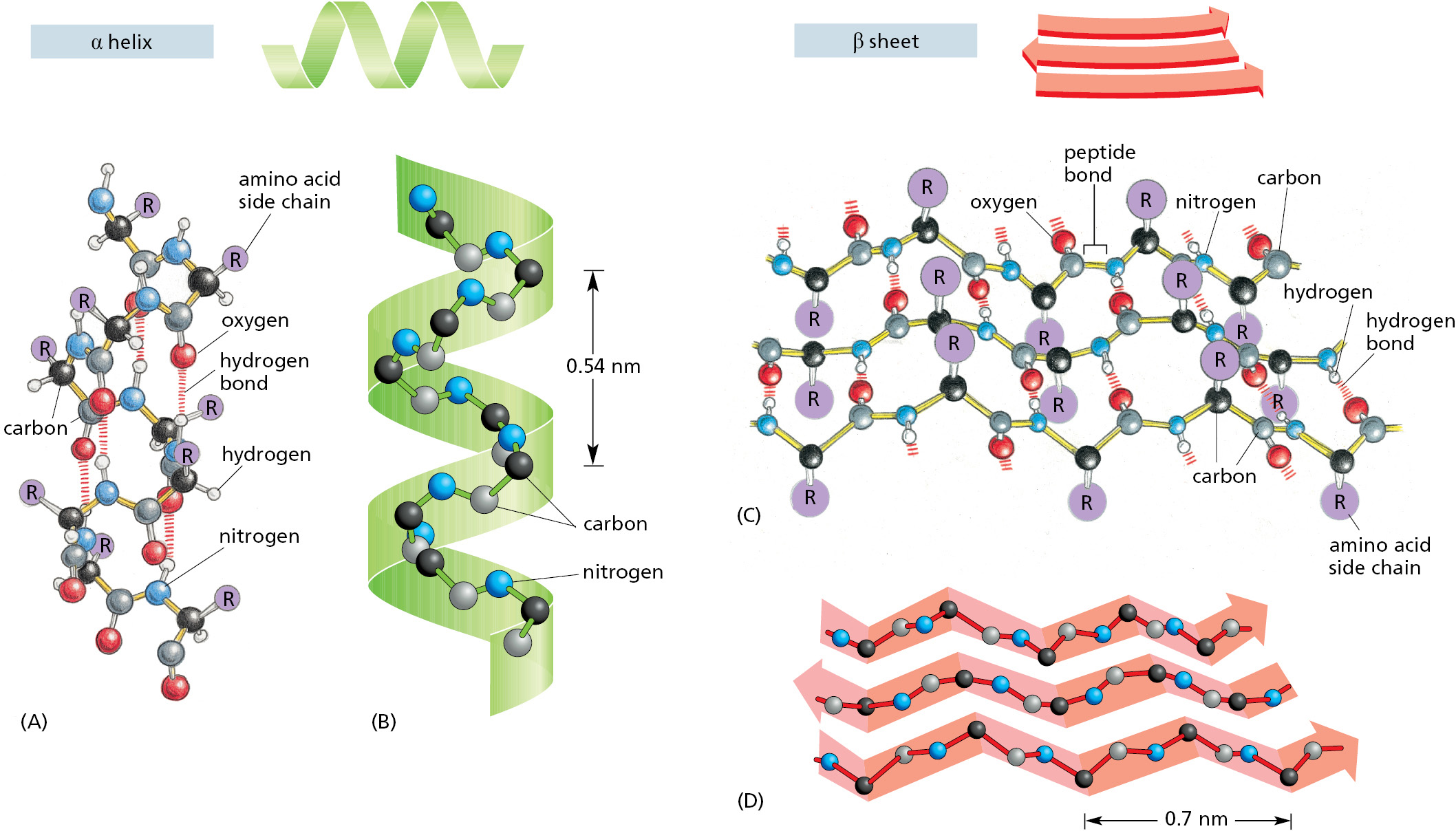
When we compare the three-dimensional structures of many different protein molecules, it becomes clear that, although the overall conformation of each protein is unique, two regular folding patterns are often found within them. Both patterns were discovered 70 years ago from studies of hair and silk. The first folding pattern to be described, called the α helix, was found in the protein α-keratin, which forms the filaments in hair. Within a year of the discovery of the α helix, a second folded structure, called a β sheet, was found in the protein fibroin, the major constituent of silk. These two patterns are common because they result from hydrogen-bonding between the N—H and C═O groups in the polypeptide backbone, without involving the side chains of the amino acids. Thus, although incompatible with some amino acid side chains, many different amino acid sequences can form them. In each case, the protein chain adopts a regular, repeating conformation. Figure 3–6 illustrates the detailed structures of these two important conformations, which in ribbon models of proteins are represented by a helical ribbon and by a set of aligned arrows, respectively.
Figure 3–6The regular conformation of the polypeptide backbone in the α helix and the β sheet. The α helix (alpha helix) is shown in (A) and (B). The N─H of every peptide bond is hydrogen-bonded to the C═O of a neighboring peptide bond located four peptide bonds away in the same chain. Note that all of the N─H groups point up in this diagram and that all of the C═O groups point down (toward the C-terminus); this gives a polarity to the helix, with the C-terminus having a partial negative and the N-terminus a partial positive charge (Movie 3.1). The β sheet (beta sheet) is shown in (C) and (D). In this example, adjacent peptide chains run in opposite (antiparallel) directions. Hydrogen-bonding between peptide bonds in different strands holds the individual polypeptide chains (strands) together in a β sheet, and the amino acid side chains in each strand alternately project above and below the plane of the sheet. By convention, when arrows are used to represent a β sheet, the arrowheads point toward the C-terminus (Movie 3.2). (A) and (C) show all the atoms in the polypeptide backbone, but the amino acid side chains are truncated and denoted by R. (It has long been a convention to use R in this way.) In contrast, (B) and (D) show only the carbon and nitrogen backbone atoms.
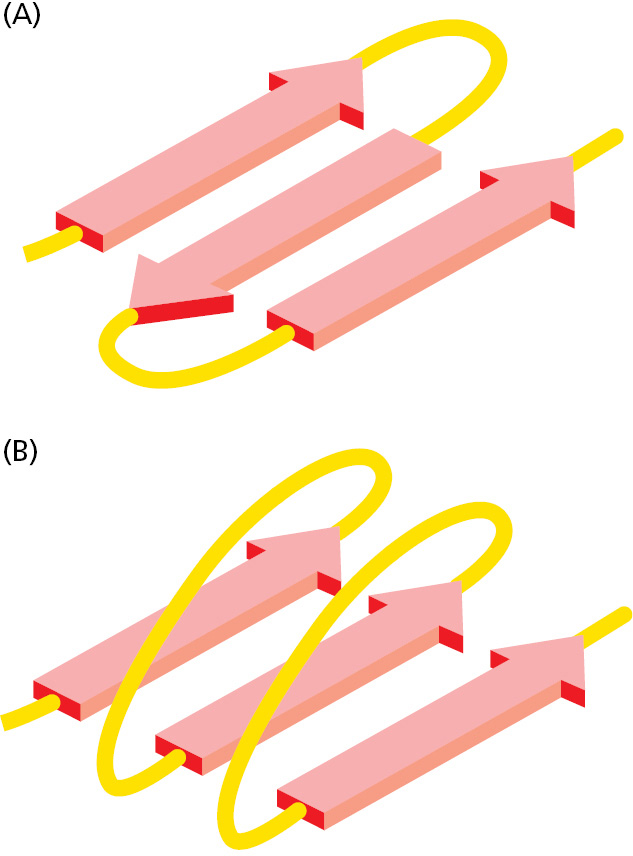
The cores of many proteins contain extensive regions of β sheet. As shown in Figure 3–7, these β sheets can form either from neighboring segments of the polypeptide backbone that run in the same orientation (parallel chains) or from a polypeptide backbone that folds back and forth upon itself, with each section of the chain running in the direction opposite to that of its immediate neighbors (antiparallel chains). Both types of β sheet produce a very rigid structure, held together by hydrogen bonds that connect the peptide bonds in neighboring chains (see Figure 3–6C).
Figure 3–7Two types of β sheet structures. (A) An antiparallel β sheet (see Figure 3–6C). (B) A parallel β sheet. Both of these structures are common in proteins.
An α helix is generated when a single polypeptide chain twists around on itself to form a rigid cylinder. A hydrogen bond forms between every fourth peptide bond, linking the C═O of one peptide bond to the N—H of another (see Figure 3–6A). This gives rise to a regular helix with a complete turn every 3.6 amino acids.
Regions of α helix are abundant in proteins located in cell membranes, such as transport proteins and receptors. As we discuss in Chapter 10, those portions of a transmembrane protein that cross the lipid bilayer usually cross as α helices composed largely of amino acids with nonpolar side chains. The polypeptide backbone, which is hydrophilic, is hydrogen-bonded to itself in the α helix and shielded from the hydrophobic lipid environment of the membrane by its protruding nonpolar side chains (see Figure 10–19).
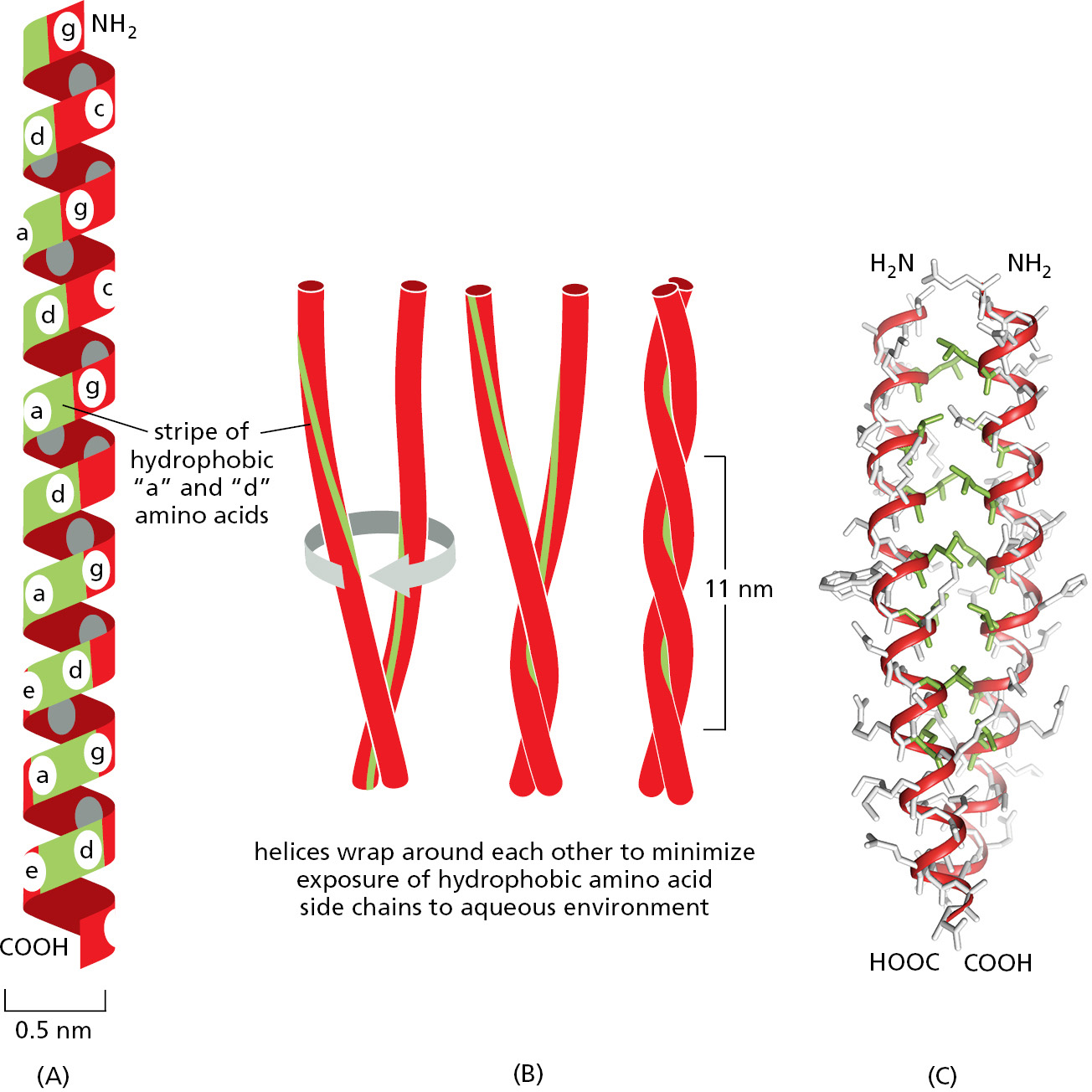
In other proteins, α helices can wrap around each other to form a particularly stable structure, known as a coiled-coil. This structure can form when the two (or in some cases, three or four) α helices have most of their nonpolar (hydrophobic) side chains on one side, so that they can twist around each other with these side chains facing inward (Figure 3–8). Long rodlike coiled-coils provide the structural framework for many elongated proteins. Examples are α-keratin, which forms the intracellular fibers that reinforce the outer layer of the skin and its appendages, and the myosin molecules responsible for muscle contraction.
Figure 3–8A coiled-coil. (A) A single α helix, with successive amino acid side chains labeled in a sevenfold sequence, “abcdefg” (from top to bottom). Amino acids “a” and “d” in such a sequence lie close together on the cylinder surface, forming a “stripe” (green) that winds slowly around the α helix. Proteins that form coiled-coils typically have nonpolar amino acids at positions “a” and “d.” Consequently, as shown in (B), the two α helices can wrap around each other with the nonpolar side chains of one α helix interacting with the nonpolar side chains of the other. (C) The atomic structure of a coiled-coil determined by x-ray crystallography. The α-helical backbone is shown in red and the nonpolar side chains in green, while the more hydrophilic amino acid side chains, shown in gray, are left exposed to the aqueous environment (Movie 3.3). Coiled-coils can also form from three α helices. (PDB code: 3NMD.)
Four Levels of Organization Are Considered to Contribute to Protein Structure
Scientists have found it useful to define four levels of organization that successively generate the structure of a protein. The first level is the protein’s amino acid sequence, which is known as its primary structure; this sequence is unique for each protein, as determined by the gene that encodes that protein. At the next level, those stretches of the polypeptide chain that form α helices and β sheets constitute the protein’s secondary structure. The full three-dimensional organization of a polypeptide chain—including its α helices, β sheets, and the many twists and turns that form between its N- and C-termini—is referred to as the protein’s tertiary structure. And finally, if a protein molecule is formed as a complex of more than one polypeptide chain, its complete conformation is designated as its quaternary structure.
Because even a small protein molecule is built from thousands of atoms linked together by precisely oriented covalent and noncovalent bonds, biologists are aided in visualizing these extremely complicated structures by computer-based three-dimensional displays. The student resource site that accompanies this book contains computer-generated images of selected proteins, which can be displayed and rotated on the screen in a variety of formats (Movie 3.4).
Protein Domains Are the Modular Units from Which Larger Proteins Are Built
Proteins come in a wide variety of shapes, and most are between 50 and 2000 amino acids long. Large proteins usually consist of a set of smaller protein domains that are joined together. A domain is a structural unit that folds more or less independently, being formed from perhaps 40 to 350 contiguous amino acids, and it is a modular unit from which larger proteins are constructed.
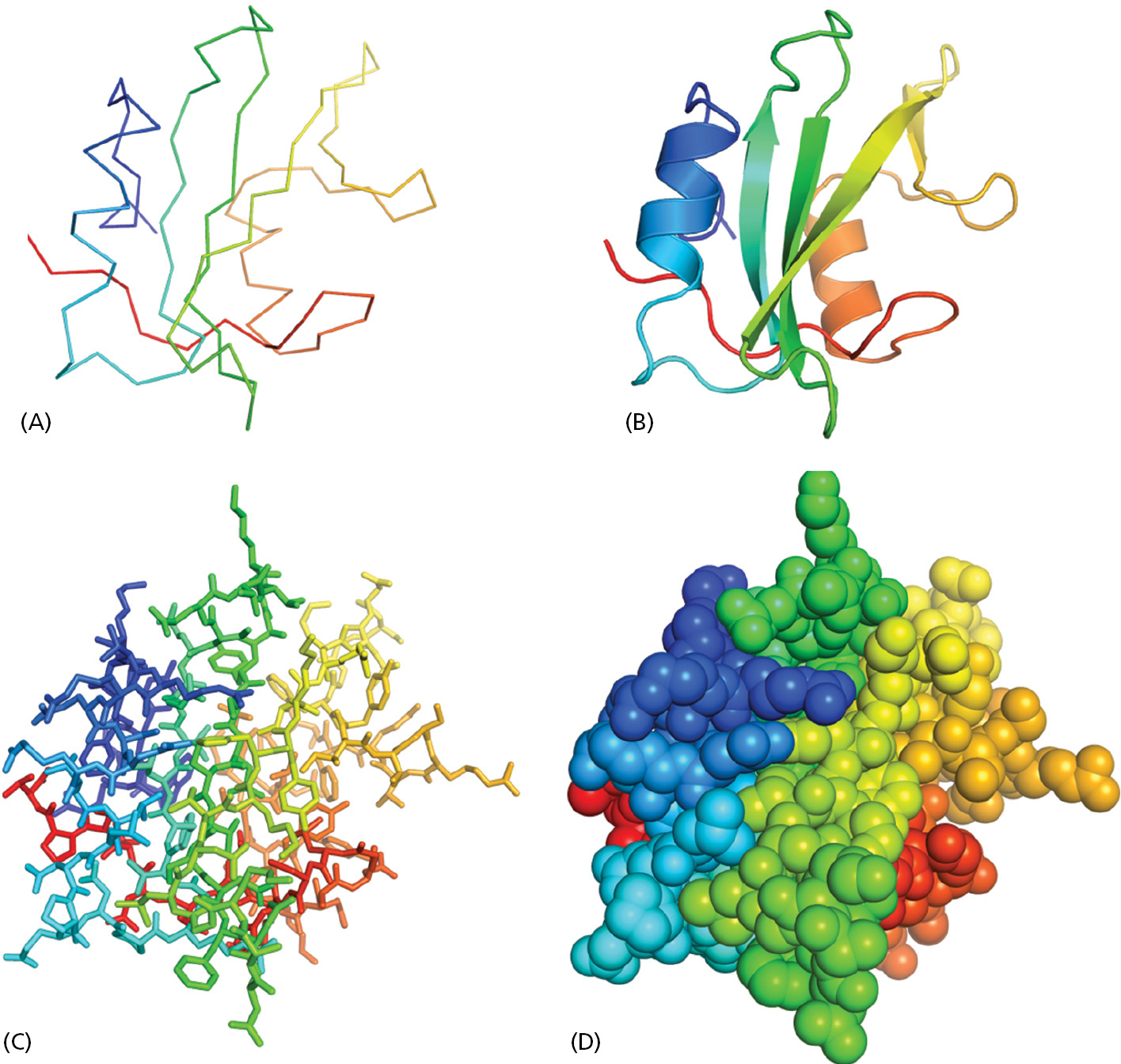
To display a protein structure in three dimensions, several different representations are conventionally used, each of which emphasizes distinct features. As an example, Figure 3–9 presents four representations of an important protein structure called the SH2 domain. The SH2 domain is present in many different proteins in eukaryotic cells, where it responds to cell signals to cause selected protein molecules to bind to each other, thereby altering cell behavior (see Chapter 15). Contributing to the tertiary structure of this domain are two α helices and a three-stranded, antiparallel β sheet, which are its critical secondary structure elements (see Figure 3–9B).
Figure 3–9Four representations that are commonly used to describe the structure of a protein. Constructed from a string of 100 amino acids, the SH2 domain is part of many different proteins. Here, its structure is displayed as (A) a polypeptide backbone model, (B) a ribbon model, (C) a wire model that includes the amino acid side chains, and (D) a space-filling model (Movie 3.4). Each image is colored in a way that allows the polypeptide chain to be followed from its N-terminus (purple) to its C-terminus (red). (PDB code: 1SHA.)
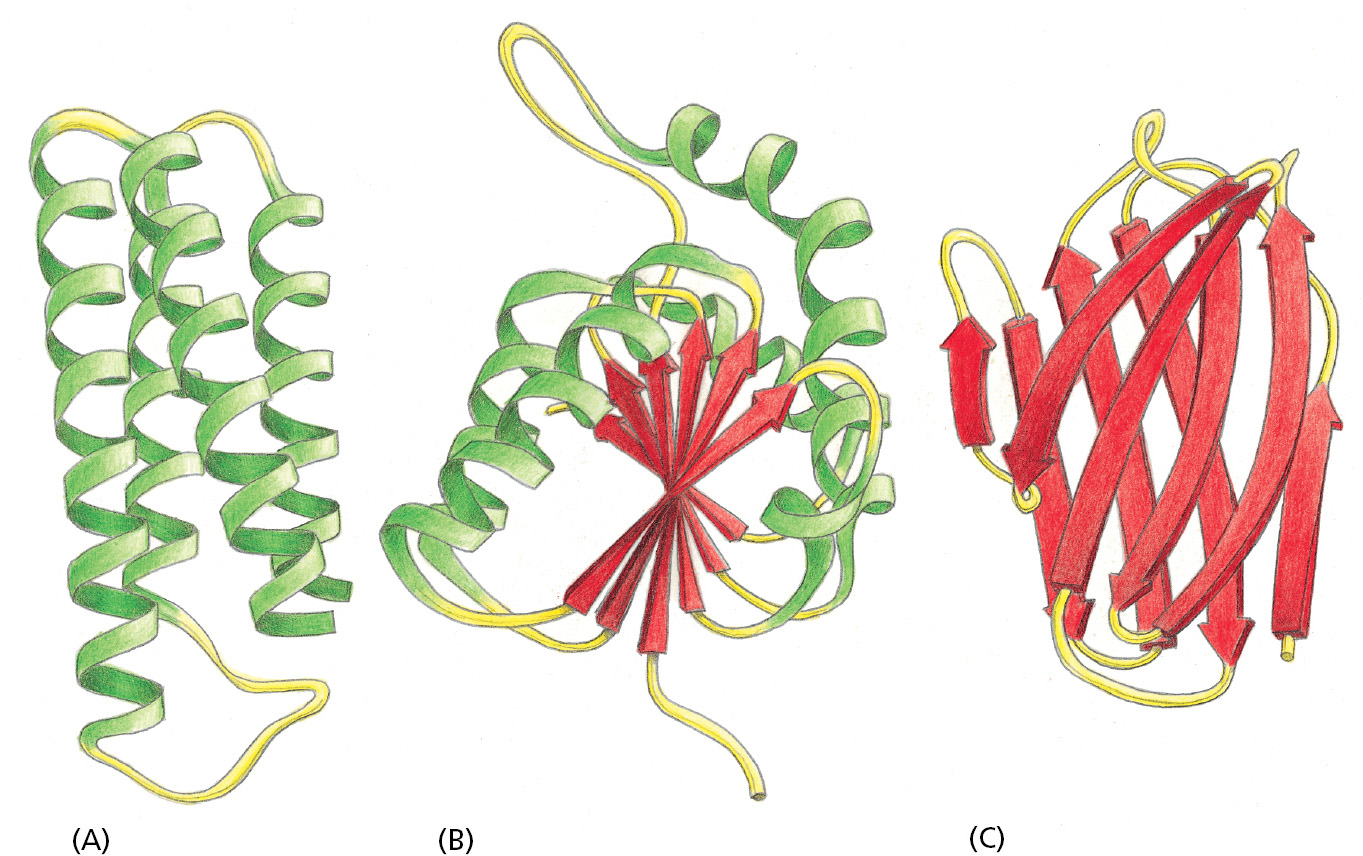
Figure 3–10 presents ribbon models of three differently organized protein domains. As these examples illustrate, the central core of a domain can be constructed from α helices, from β sheets, or from various combinations of these two fundamental folding elements.
Figure 3–10Ribbon models of three different protein domains. (A) Cytochrome b562, a single-domain protein involved in electron transport in mitochondria. This protein is composed almost entirely of α helices. (B) The NAD-binding domain of the enzyme lactate dehydrogenase, which is composed of a mixture of α helices and parallel β sheets. (C) The variable domain of an immunoglobulin (antibody) light chain, composed of a sandwich of two antiparallel β sheets. In these examples, the α helices are shown in green, while strands organized as β sheets are denoted by red arrows. Note how the polypeptide chain generally traverses back and forth across the entire domain, making sharp turns (Movie 3.5) only at the protein surface. It is the protruding loop regions (yellow) that often form the binding sites for other molecules.
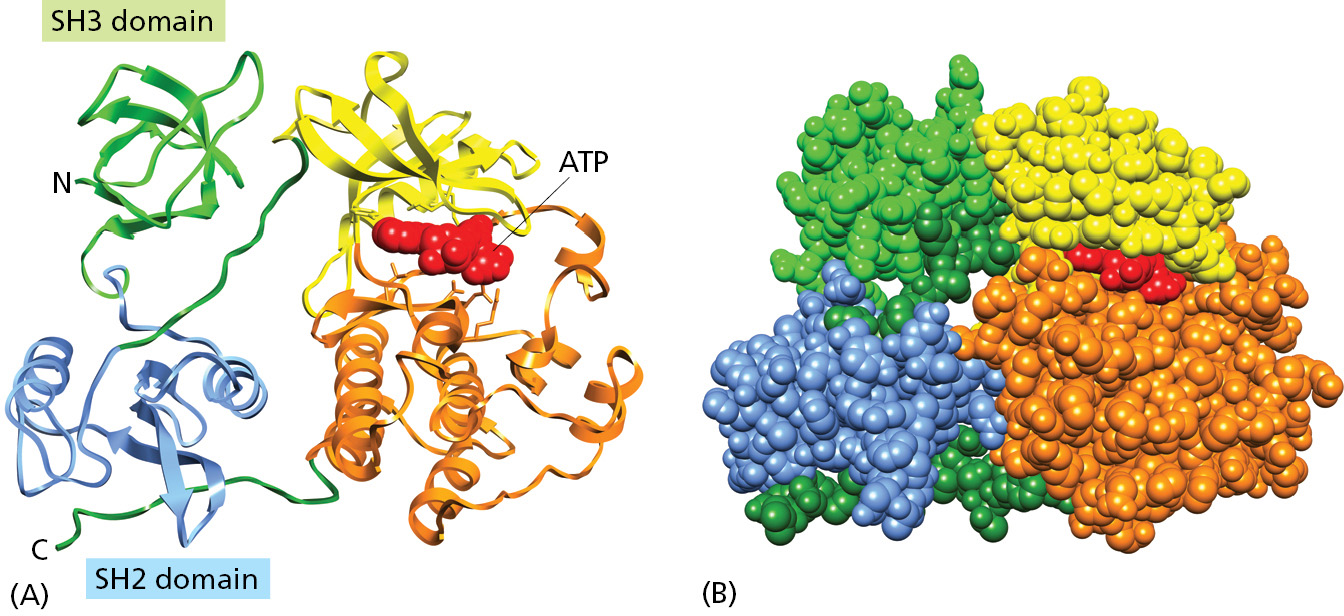
The different domains of a protein are often associated with different functions. Figure 3–11 shows an example—the Src protein kinase, which functions in signaling pathways inside vertebrate cells (Src is pronounced “sarc”). This protein is considered to have three domains: its SH2 and SH3 domains have regulatory roles—responding to signals that turn the kinase on and off—while its C-terminal domain is responsible for the kinase catalytic activity. Later in the chapter, we shall return to this protein to explain how proteins can form molecular switches that transmit information throughout cells.
Figure 3–11A protein formed from multiple domains. In the Src protein shown, a C-terminal domain with two lobes (yellow and orange) forms the core protein kinase enzyme, while its SH2 and SH3 domains perform regulatory functions. Note that both the SH2 and SH3 domains derive their names from this protein, being abbreviations for “Src homology 2” and “Src homology 3,” respectively. (A) A ribbon model, with ATP substrate in red. (B) A space-filling model, with ATP substrate in red. Note that the site that binds ATP is positioned at the interface of the two lobes that form the kinase domain. The human genome encodes about 300 different SH3 domains and 120 SH2 domains. The structure of the SH2 domain was illustrated in Figure 3–9. (PDB code: 2SRC.)
Proteins Also Contain Unstructured Regions
The smallest protein molecules contain only a single domain, whereas larger proteins can contain several dozen domains, often connected to each other by short, relatively unstructured lengths of polypeptide chain that can act as flexible hinges between domains. The ubiquity of such intrinsically disordered sequences, which continually bend and flex due to thermal buffeting, became appreciated only after bioinformatics methods were developed that could recognize them from their amino acid sequences. Current estimates suggest that a third of all eukaryotic proteins also possess longer, intrinsically disordered regions (IDRs)—greater than 30 amino acids in length—in their polypeptide chains. These intrinsically disordered regions can be very long, and they have important functions in cells, as discussed later in this chapter.
All Protein Structures Are Dynamic, Interconverting Rapidly Between an Ensemble of Closely Related Conformations Because of Thermal Energy
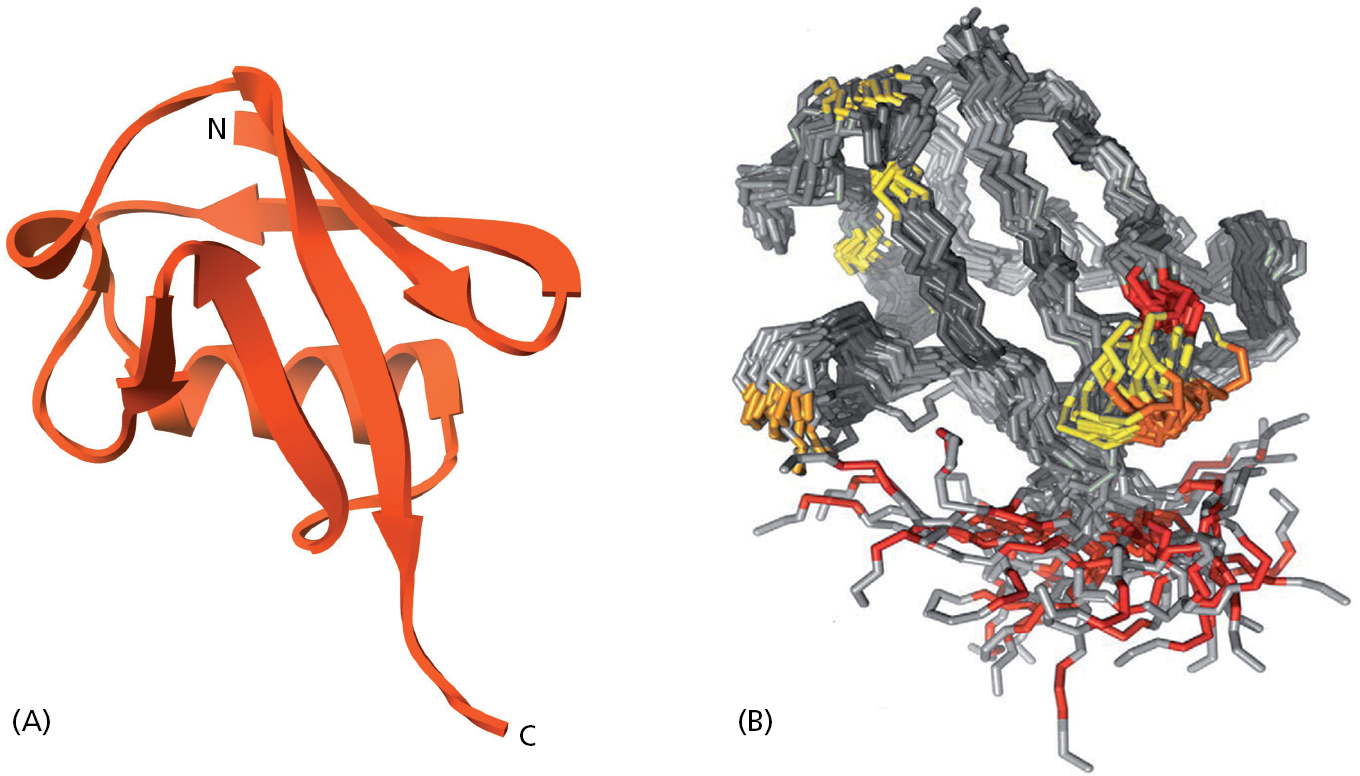
Even though a protein has folded into a conformation of lowest free energy, this conformation is always being subjected to thermal bombardment from the Brownian motions of the many molecules that constantly collide with it. Thus the atoms in the protein are always moving, which causes neighboring regions of the protein to oscillate in concerted ways. These motions can now be precisely traced using special NMR techniques, as illustrated in Figure 3–12 for the small protein ubiquitin.
Figure 3–12A folded protein molecule exists as an ensemble of closely related substructures, or conformers, as displayed here for ubiquitin. (A) A ribbon model that displays the structure of ubiquitin. Ubiquitin is a small protein widely used in cells, often being covalently attached to larger proteins, as described in Chapters 6 and 15. (B) In this diagram, a set of backbone conformations determined for ubiquitin has been overlaid to reveal regions that rapidly transition between different substructures. Superimposed on these structures are the rates of motion of the protein’s atoms, as observed in NMR residual dipolar coupling experiments. A color code has been used to indicate the magnitude of these rates, which are largest for red, with orange and yellow also being high. (A, PDB code 1UBI; B, from O.F. Lange et al., Science 320:1471–1475, 2008. With permission from AAAS.)
From recent studies combining many types of analyses, we know that protein function exploits these rapid fluctuations—as when a loop on the surface of a protein flips out to expose a binding site for a second molecule. In fact, the function of a protein is generally dependent on that protein’s dynamic character, as we explain later when we discuss protein function in detail.
Function Has Selected for a Tiny Fraction of the Many Possible Polypeptide Chains
Because each of the 20 amino acids is chemically distinct and each can, in principle, occur at any position in a protein chain, there are 20 × 20 × 20 × 20 = 160,000 different possible polypeptide chains four amino acids long, or 20n different possible polypeptide chains n amino acids long. For a typical protein length of about 300 amino acids, a cell could theoretically make more than 10390 (20300) different polypeptide chains. This is such an enormous number that to produce just one molecule of each kind would require many more atoms than exist in the universe.
Only a very small fraction of this vast set of conceivable polypeptide chains would adopt a stable three-dimensional conformation—by some estimates, less than one in a billion. And yet the majority of proteins present in cells do adopt unique and stable conformations. How is this possible? The answer lies in natural selection. A protein with an unpredictably variable structure and biochemical activity is unlikely to help the survival of a cell that contains it. Such proteins would therefore have been eliminated by natural selection through the enormously long trial-and-error process that underlies biological evolution.
Because evolution has selected for protein function in living organisms, present-day proteins have chemical properties that enable the protein to perform a particular catalytic or structural function in the cell. Proteins are so precisely built that the change of even a few atoms in one amino acid can sometimes disrupt the structure of the whole molecule so severely that all function is lost. And, as discussed later in this chapter, when certain rare protein misfolding accidents occur, the results can be disastrous for the organisms that contain them.
Proteins Can Be Classified into Many Families
Once a protein had evolved that folded up into a stable conformation with useful properties, its structure was often modified during evolution to enable it to perform new functions. As we will discuss in Chapter 4, this process has been greatly accelerated by genetic mechanisms that duplicate genes accidentally, which allows gene copies to evolve independently to perform new functions. Because this type of event occurred frequently in the past, present-day proteins can be grouped into protein families, each family member having an amino acid sequence and a three-dimensional conformation that resemble those of the other family members.
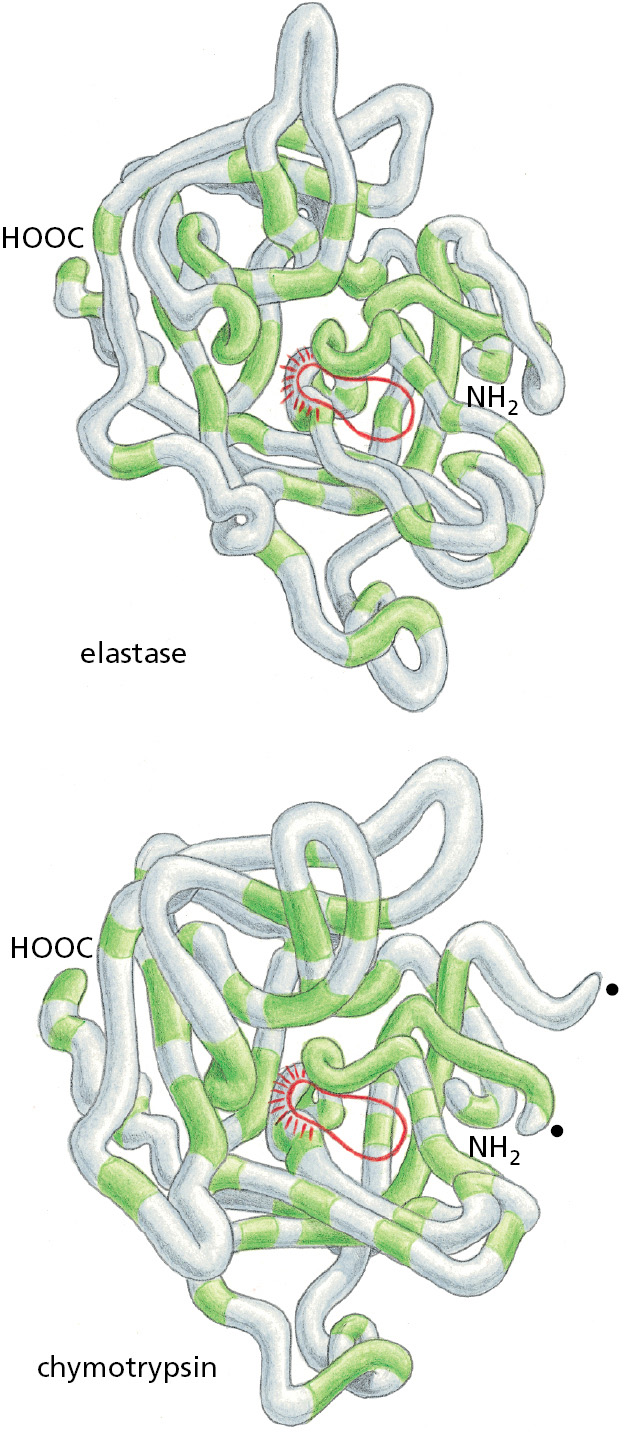
Consider, for example, the serine proteases, a large family of protein-cleaving (proteolytic) enzymes that includes the digestive enzymes chymotrypsin, trypsin, and elastase, as well as several proteases involved in blood clotting. When the protease portions of any two of these enzymes are compared, parts of their amino acid sequences are found to match. The similarity of their three-dimensional conformations is even more striking: most of the detailed twists and turns in their polypeptide chains, which are several hundred amino acids long, are virtually identical (Figure 3–13). The many different serine proteases nevertheless have distinct enzymatic activities, each cleaving different proteins or the peptide bonds between different types of amino acids. Each therefore performs a distinct function in an organism.
Figure 3–13A comparison of the conformations of two serine proteases. The backbone conformations of elastase and chymotrypsin. Although only those amino acids in the polypeptide chain shaded in green are the same in the two proteins, the two conformations are very similar nearly everywhere. The active site of each enzyme is circled in red; this is where the peptide bonds of the proteins that serve as substrates are bound and cleaved by hydrolysis. The serine proteases derive their name from the amino acid serine, whose side chain is part of the active site of each enzyme and directly participates in the cleavage reaction. The two dots on the right side of the chymotrypsin molecule mark the new ends created when this enzyme cuts its own backbone.
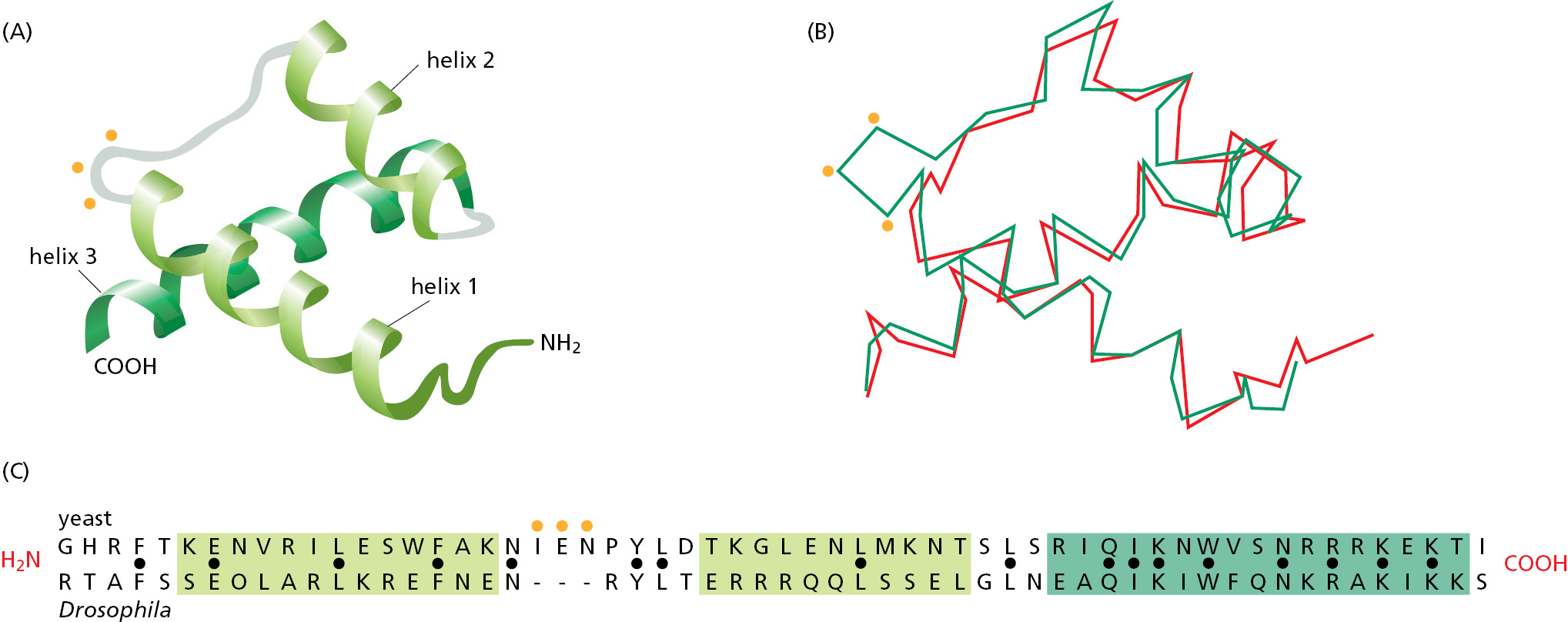
The story we have told for the serine proteases could be repeated for hundreds of other protein families. In general, the structure of the different members of a protein family has been more highly conserved than has the amino acid sequence. In many cases, the amino acid sequences have diverged so far that we cannot be certain of a family relationship between two proteins without determining their three-dimensional structures. The yeast α2 protein and the Drosophila engrailed protein, for example, are both transcription regulatory proteins in the homeodomain family (discussed in Chapter 7). Because they are identical in only 17 of the 60 amino acids of their homeodomain, their relationship became certain only by comparing their three-dimensional structures (Figure 3–14). Many similar examples show that two proteins with more than 25% identity in their amino acid sequences usually share the same overall structure.
Figure 3–14A comparison of a class of DNA-binding domains, called homeodomains, in a pair of proteins from two organisms separated by more than a billion years of evolution. (A) A ribbon model of the structure common to both proteins. (B) A trace of the α-carbon positions. The three-dimensional structures shown were determined by x-ray crystallography for the yeast α2 protein (green) and the Drosophila engrailed protein (red). (C) A comparison of amino acid sequences for the region of the proteins shown in A and B. Black dots mark sites with identical amino acids. Green shading has been used to mark the three α helices shown in A. Orange dots indicate the position of a three-amino-acid insert in the α2 protein. (Adapted from C. Wolberger et al., Cell 67:517–528, 1991.)
The various members of a large protein family often have distinct functions. Mutation is a random process. Some of the amino acid changes that make family members different were selected in the course of evolution because they resulted in useful changes in biological activity; these give the individual family members the different functional properties they have today. Other amino acid changes were effectively “neutral,” having neither a beneficial nor a damaging effect on the basic structure and function of the protein. In addition, because mutation is random, there must also have been many deleterious changes that altered the three-dimensional structure of these proteins sufficiently to make them useless. Such faulty proteins would have been readily lost during evolution.
Protein families are readily recognized when the genome of any organism is sequenced; for example, the determination of the DNA sequence for the entire human genome has revealed that we contain about 20,000 protein-coding genes. Through sequence comparisons, we can assign the products of more than half of our protein-coding genes to known protein structures belonging to more than 500 different protein families. Most of the proteins in each family have evolved to perform somewhat different functions, as for the enzymes elastase and chymotrypsin illustrated previously in Figure 3–13. These family members are sometimes called paralogs to distinguish them from orthologs—those evolutionarily related proteins that have the same function in different organisms (such as the mouse elastase and human elastase enzymes).
The current database of known protein sequences contains more than 100 million entries, and it is growing very rapidly as more and more genomes are sequenced—revealing huge numbers of new genes that encode proteins. The encoded polypeptides range widely in size, from 6 amino acids to a gigantic protein of 34,000 amino acids (titin, a structural protein in muscle).
As described in Chapters 8 and 9, because of the powerful techniques of x-ray crystallography, nuclear magnetic resonance (NMR), and cryo-electron microscopy, we now know the three-dimensional shapes, or conformations, of more than 100,000 of these proteins. By carefully comparing the conformations of these proteins, structural biologists (that is, experts on the structure of biological molecules) have concluded that there are a limited number of ways in which protein domains usually fold up in nature—estimated to be about 2000, if we consider all organisms. For most of these so-called protein folds, representative structures have been determined.
Protein comparisons are important because related structures often imply related functions. Many years of experimentation can be saved by discovering that a new protein has an amino acid sequence similarity with a protein of known function. Such sequence relationships, for example, first indicated that certain genes that cause mammalian cells to become cancerous encode protein kinases (discussed in Chapter 20).
Some Protein Domains Are Found in Many Different Proteins
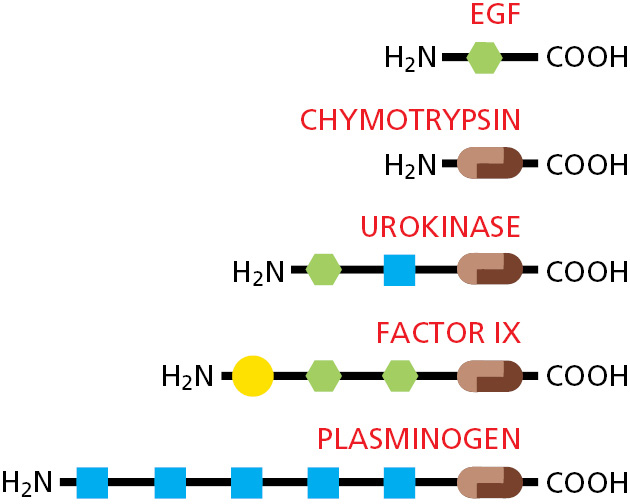
As previously stated, most proteins are composed of a series of protein domains in which different regions of the polypeptide chain fold independently to form compact structures. Such multidomain proteins are believed to have originated from the accidental joining of the DNA sequences that encode each domain, creating a new gene. In an evolutionary process called domain shuffling, many large proteins have evolved through the joining of preexisting domains in new combinations (Figure 3–15). Novel binding surfaces have often been created at the juxtaposition of domains, and many of the functional sites where proteins bind to small molecules are found to be located there.
Figure 3–15Domain shuffling. An extensive shuffling of blocks of protein sequence (protein domains) has occurred during protein evolution. Those portions of a protein denoted by the same shape and color in this diagram are evolutionarily related. Serine proteases such as chymotrypsin are formed from two domains (brown). In the three other proteases shown, which are highly regulated and more specialized, these two protease domains are connected to one or more domains that are similar to domains found in epidermal growth factor (EGF; green), to a calcium-binding protein (yellow), or to a kringle domain (blue). Chymotrypsin is illustrated in Figure 3–13.
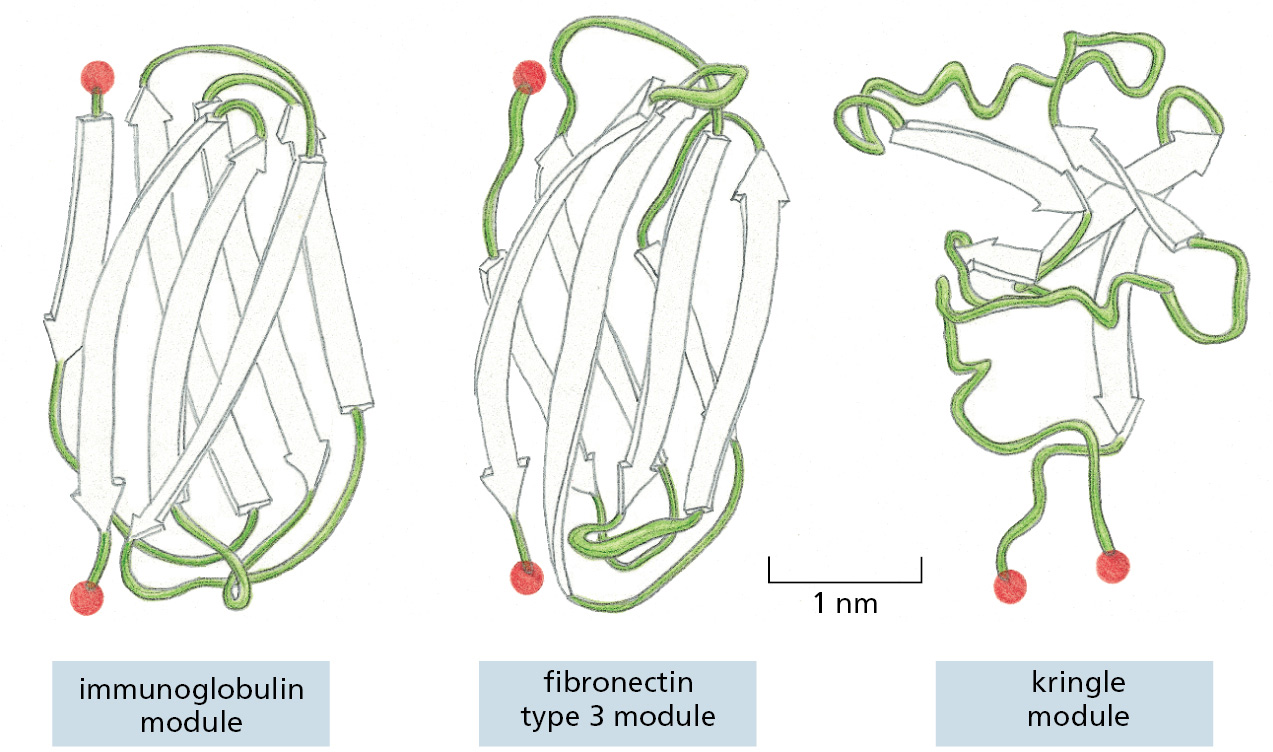
A subset of protein domains has been especially mobile during evolution; these seem to have particularly versatile structures and are sometimes referred to as protein modules. The structure of one such module, the SH2 domain, was featured in Figure 3–9. Three other abundant protein domains are illustrated in Figure 3–16.
Figure 3–16The three-dimensional structures of three commonly used protein domains. In these ribbon diagrams, β-sheet strands are shown as arrows, and the N- and C-termini are indicated by red spheres. Many more such “protein modules” exist in nature. (Adapted from D.J. Leahy et al., Science 258:987–991, 1992. With permission from AAAS.)
Each of these three domains has a stable core structure formed from strands of β sheets, from which less-ordered loops of polypeptide chain protrude. The loops are ideally situated to form binding sites for other molecules, as most clearly demonstrated for the immunoglobulin fold, which forms the basis for antibody molecules. Such β sheet–based domains may have achieved their evolutionary success because they provide a convenient framework for the generation of new binding sites for ligands, requiring only small changes to their protruding loops (see Figure 3–40).
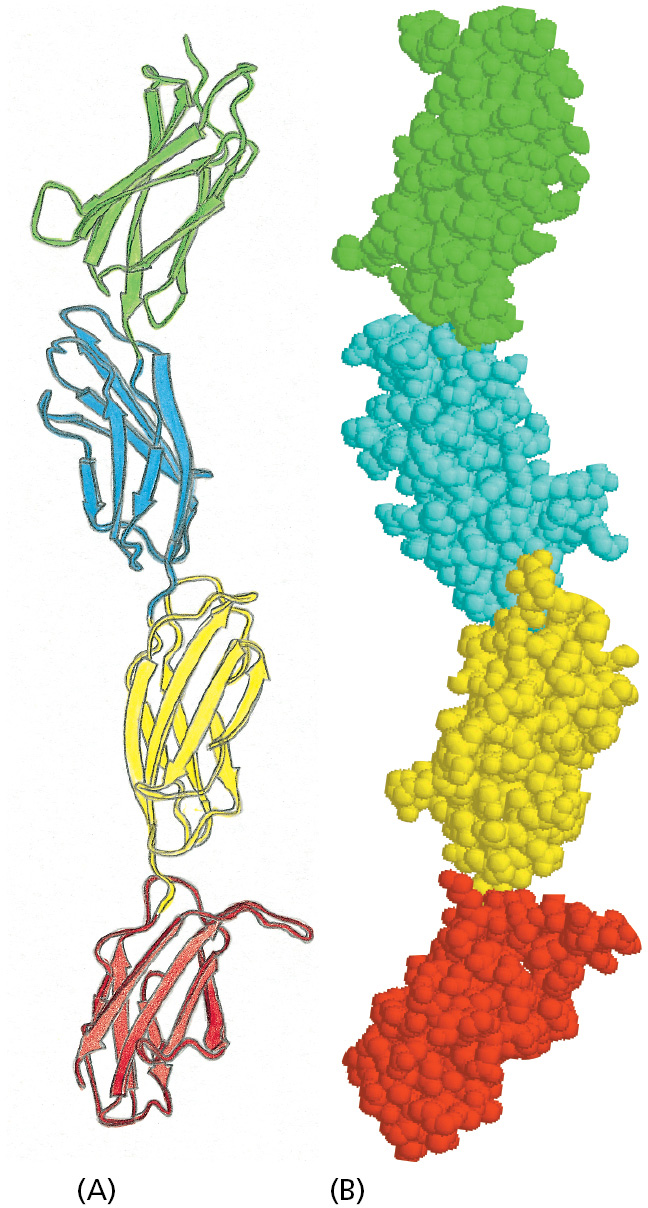
A second feature of these protein domains that explains their utility is the ease with which they can be integrated into other proteins. Two of the three domains illustrated in Figure 3–16 have their N- and C-terminal ends at opposite poles of the domain. When the DNA encoding such a domain undergoes tandem duplication, which is not unusual in the evolution of genomes (discussed in Chapter 4), the duplicated domains with this in-line arrangement can be readily linked in series to form extended structures—either with themselves or with other in-line domains (Figure 3–17). Stiff extended structures composed of a series of domains are especially common in extracellular matrix molecules and in the extracellular portions of cell-surface receptor proteins. Other frequently used domains, including the SH2 domain and the kringle domain in Figure 3–16, are of a plug-in type, with their N- and C-termini close together. After genomic rearrangements, such domains are usually accommodated as an insertion into a loop region of a second protein.
Figure 3–17An extended structure formedfrom a series of protein domains. Four fibronectin type 3 domains (see Figure 3–16) from the extracellular matrix molecule fibronectin are illustrated in (A) ribbon and (B) space-filling models. (Adapted from D.J. Leahy et al., Cell 84:155–164, 1996.)
A comparison of the relative frequency of domain utilization in different eukaryotes reveals that for many common domains, such as protein kinases, this frequency is similar in organisms as diverse as yeast, plants, worms, flies, and humans. But there are some notable exceptions, such as the major histocompatibility complex (MHC) antigen-recognition domain (see Figure 24–36) that is present in 57 copies in humans, but absent in the other four organisms just mentioned. Domains such as these have specialized functions that are not shared with the other eukaryotes; they are assumed to have been strongly selected for during recent evolution to produce the multiple copies observed.
The Human Genome Encodes a Complex Set of Proteins, Revealing That Much Remains Unknown
The result of sequencing the human genome has been surprising, because it reveals that our chromosomes contain only about 20,000 protein-coding genes. On the basis of this number alone, we would appear to be no more complex than the tiny mustard weed, Arabidopsis, and only about 1.3-fold more complex than a nematode worm. The genome sequences also reveal that vertebrates have inherited nearly all of their protein domains from invertebrates—with only 7% of identified human domains being vertebrate specific.
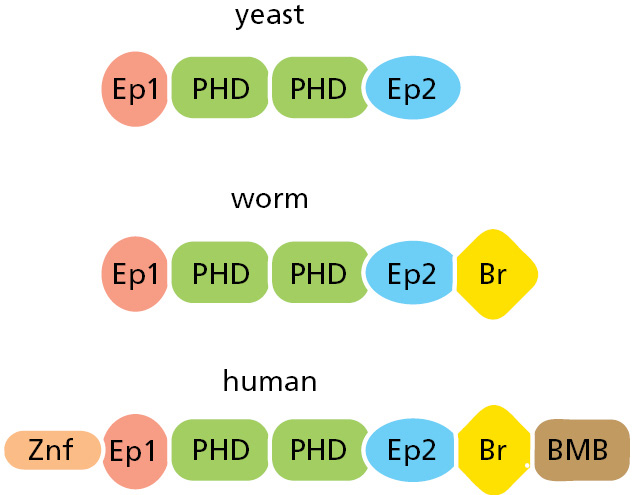
Figure 3–18Domains in a group of evolutionarily related proteins that have a similar function. In general, there is a tendency for the proteins in more complex organisms, such as humans, to contain additional domains compared to a less complex organism such as yeast—as is the case for the DNA-binding protein compared here.
Each of our proteins is on average more complicated, however (Figure 3–18). Domain shuffling during vertebrate evolution has given rise to many novel combinations of protein domains, with the result that there are nearly twice as many combinations of domains found in human proteins as in a worm or a fly. This extra variety in our proteins greatly increases the range of protein–protein interactions possible, but how it contributes to making us human is not known.
The complexity of living organisms is staggering, and it is quite sobering to note that we currently lack even the tiniest hint of what the function might be for more than 10,000 of the proteins that have been identified through examining the human genome. There are certainly enormous challenges ahead for the next generation of cell biologists, with no shortage of fascinating mysteries to solve.
Protein Molecules Often Contain More Than One Polypeptide Chain
The same weak noncovalent bonds that enable a protein chain to fold into a specific conformation also allow proteins to bind to each other to produce larger structures in the cell. Any region of a protein’s surface that can interact with another molecule through sets of noncovalent bonds is called a binding site. A protein can contain binding sites for various large and small molecules. If a binding site recognizes the surface of a second protein, the tight binding of two folded polypeptide chains at this site creates a larger protein molecule with a precisely defined geometry. Each polypeptide chain in such a protein is called a protein subunit. And the precise way that these subunits are arranged creates the protein’s quaternary structure—as introduced previously.
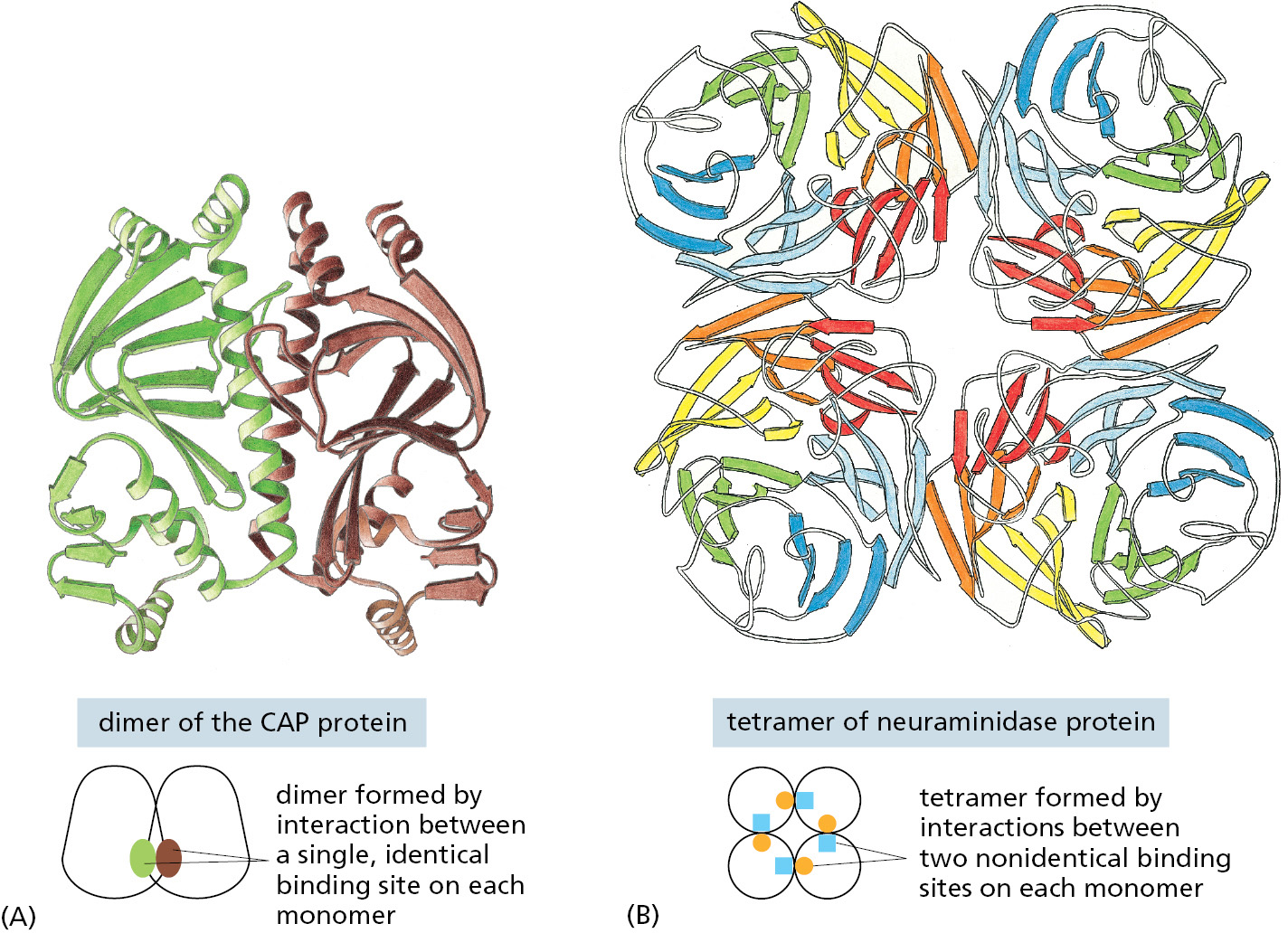
In the simplest case, two identical, folded polypeptide chains form a symmetrical complex of two protein subunits (called a dimer) that is held together by interactions between two identical binding sites. (Figure 3–19A). Symmetrical protein complexes that are formed from more than two copies of the same polypeptide chain are also commonly found in cells (Figure 3–19B).
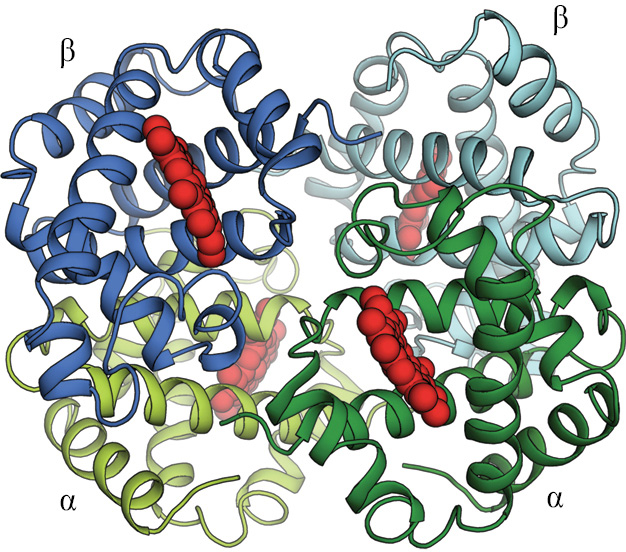
Figure 3–19Many protein molecules contain multiple copies of the same protein subunit. (A) A symmetrical dimer. The CAP protein, a bacterial transcription regulatory protein, is a complex of two identical polypeptide chains. (B) A symmetrical homotetramer. The enzyme neuraminidase exists as a ring of four identical polypeptide chains. For both A and B, a small schematic below the structure emphasizes how the repeated use of the same binding interaction forms the structure. In A, the use of the same binding site on each monomer (represented by brown and green ovals) causes the formation of a symmetrical dimer. In B, a pair of nonidentical binding sites (represented by orange circles and blue squares) causes the formation of a symmetrical tetramer.Figure 3–20Hemoglobin is a protein formed as a symmetrical assembly using two each of two different subunits. This abundant, oxygen-carrying protein in red blood cells contains two copies of α-globin (green) and two copies of β-globin (blue). Each of these four polypeptide chains contains a heme molecule (red), which is the site that binds oxygen (O2). Thus, each molecule of hemoglobin carries four molecules of oxygen. (PDB code: 2DHB.)
Many other proteins contain two or more types of polypeptide chains. Hemoglobin, the protein that carries oxygen in red blood cells, contains two identical α-globin subunits and two identical β-globin subunits, symmetrically arranged (Figure 3–20). Such multisubunit proteins can be very large(Movie 3.6).
Some Globular Proteins Form Long Helical Filaments
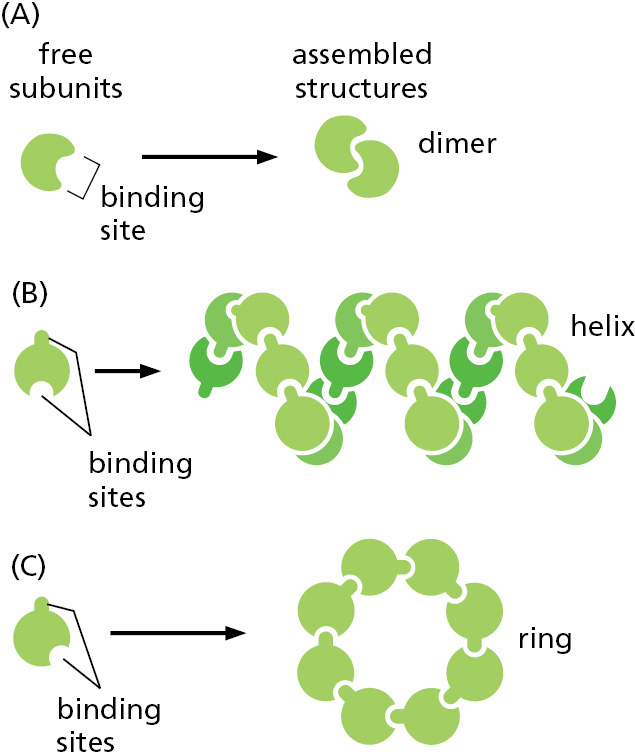
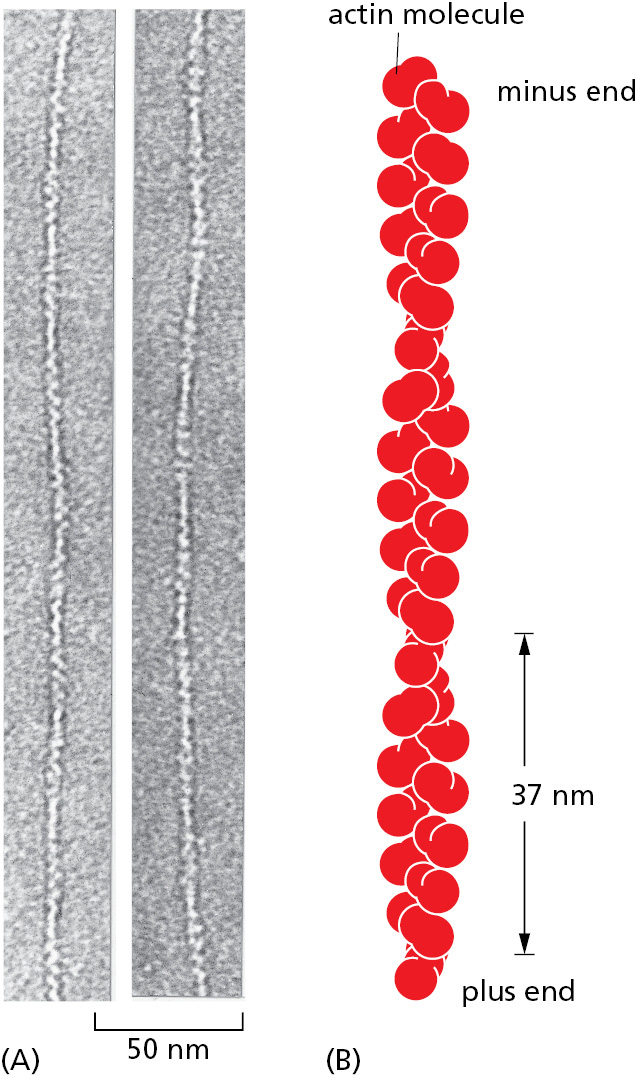
The proteins that we have discussed so far are globular proteins, in which the polypeptide chain folds up into a compact shape like a ball with an irregular surface. Some of these protein molecules can nevertheless assemble to form filaments that may span the entire length of a cell. Most simply, a long chain of identical protein molecules can be constructed if each molecule has a binding site complementary to another region of the surface of the same molecule (Figure 3–21). An actin filament, for example, is a long helical structure produced from many molecules of the protein actin (Figure 3–22). Actin is a globular protein that is very abundant in eukaryotic cells, where it forms one of the major filament systems of the cytoskeleton (discussed in Chapter 16).
Figure 3–21Protein assemblies. (A) A protein with just one binding site can form a dimer with another identical protein. (B) Identical proteins with two different binding sites often form a long helical filament. (C) If the two binding sites are disposed appropriately in relation to each other, the protein subunits may form a closed ring instead of a helix. (For an example of A, see Figure 3–19A; for an example of B, see Figure 3–22; for an example of C, see Figure 14–32.)Figure 3–22Globular actin monomers assemble to produce an actin filament. (A) Transmission electron micrographs of negatively stained actin filaments. (B) The helical arrangement of actin molecules in an actin filament. (A, courtesy of Roger Craig.)
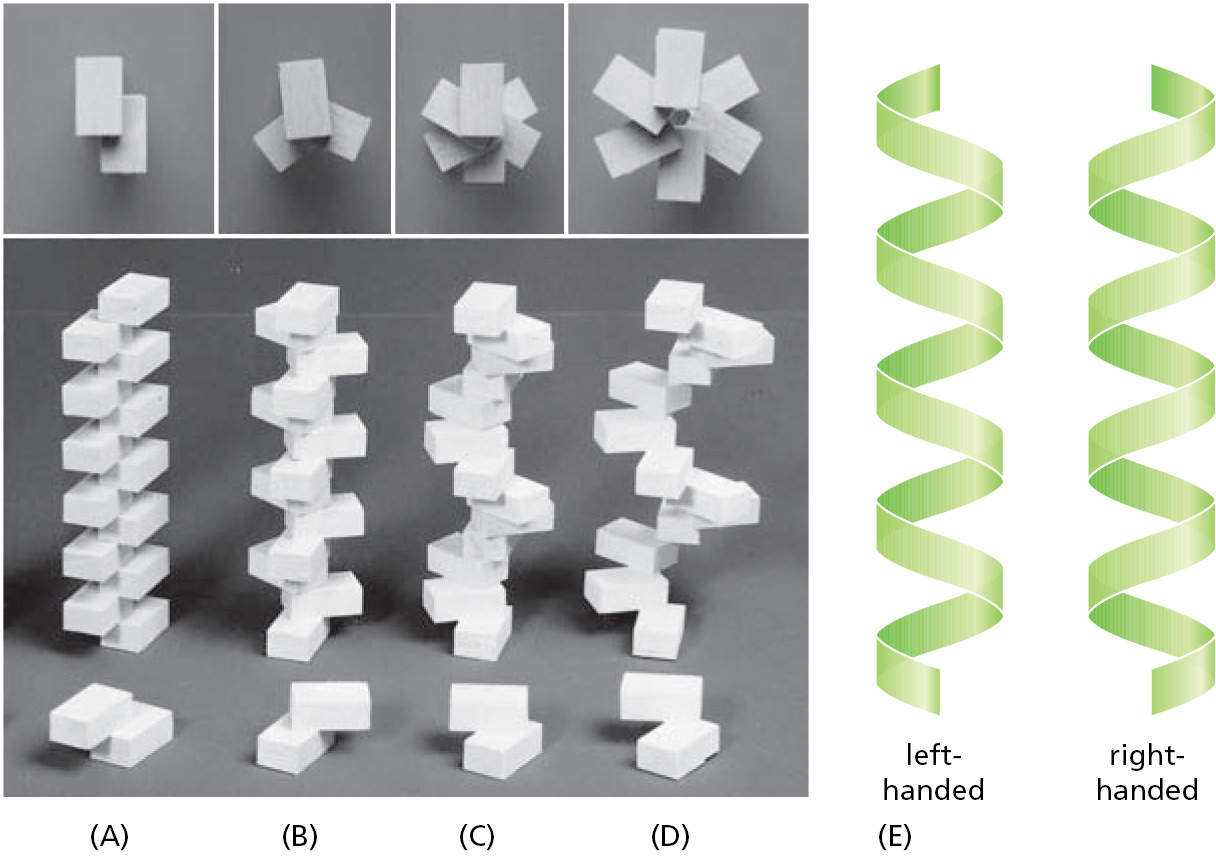
We will encounter many helical structures in this book. Why is a helix such a common structure in biology? As we have seen, biological structures are often formed by linking similar subunits into long, repetitive chains. If all the subunits are identical, the neighboring subunits in the chain can often fit together in only one way, adjusting their relative positions to minimize the free energy of the contact between them. As a result, each subunit is positioned in exactly the same way in relation to the next, so that subunit 3 fits onto subunit 2 in the same way that subunit 2 fits onto subunit 1, and so on. Because it is very rare for subunits to join up in a straight line, this arrangement generally results in a helix—a regular structure that resembles a spiral staircase, as illustrated in Figure 3–23. Depending on the twist of the staircase, a helix is said to be either right-handed or left-handed (see Figure 3–23E). Handedness is not affected by turning the helix upside down, but it is reversed if the helix is reflected in the mirror.
Figure 3–23Some properties of a helix. (A–D) A helix forms when a series of subunits (here represented by rectangular bricks) bind to each other in a regular way. At the top, each of these helices is viewed from directly above the helix and seen to have two (A), three (B), and six (C and D) subunits per helical turn. Note that the helix in D has a wider path than that in C but the same number of subunits per turn. (E) As discussed in the text, a helix can be either right-handed or left-handed. As a reference, it is useful to remember that standard metal screws, which insert when turned clockwise, are right-handed. Note that a helix retains the same handedness when it is turned upside down.
The observation that helices occur commonly in biological structures holds true whether the subunits are small molecules linked together by covalent bonds (for example, the amino acids in an α helix) or large protein molecules that are linked by noncovalent forces (for example, the actin molecules in actin filaments). This is not surprising. A helix is an unexceptional structure, and it is generated simply by placing many similar subunits next to each other, each in the same strictly repeated relationship to the one before; that is, with a fixed rotation followed by a fixed translation along the helix axis.
Protein Molecules Can Have Elongated, Fibrous Shapes
Enzymes tend to be globular proteins: even though many are large and complicated, with multiple subunits, most have an overall rounded shape. In Figure 3–22, we saw that a globular protein can associate to form long filaments. But some functions require that an individual protein molecule span a large distance. These fibrous proteins generally have a relatively simple, elongated three-dimensional structure.
One large family of intracellular fibrous proteins consists of α-keratin, introduced when we described the α helix. Keratin filaments are extremely stable and are the main component in long-lived structures such as hair, horn, and nails. An α-keratin molecule is a dimer of two identical subunits, with the long α helices of each subunit forming a coiled-coil (see Figure 3–8). The coiled-coil regions are capped at each end by globular domains containing binding sites. This enables this type of protein to assemble into ropelike intermediate filaments—an important component of the cytoskeleton that creates the cell’s internal structural framework (see Figure 16–62).
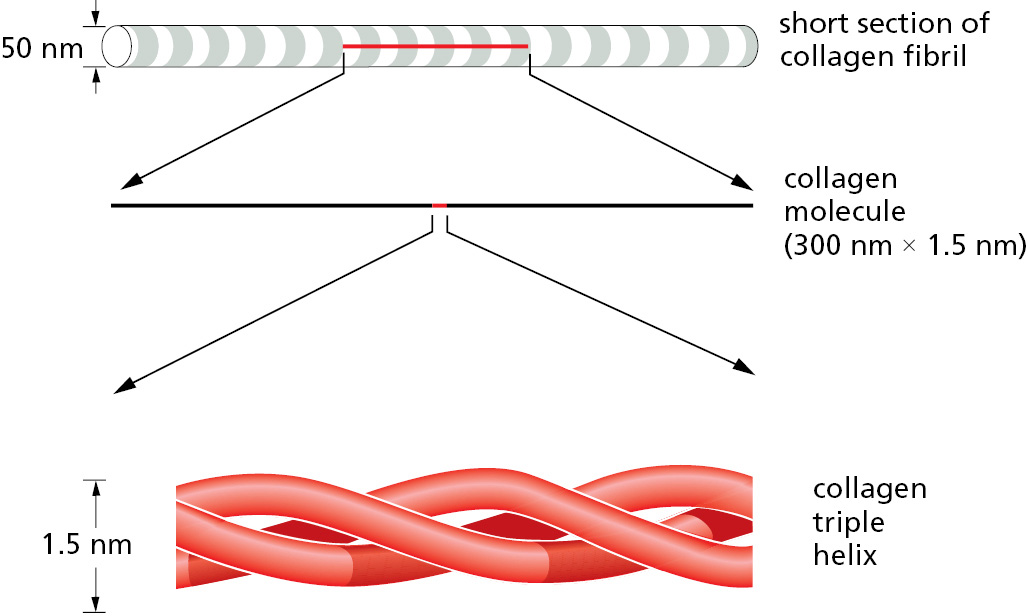
Fibrous proteins are especially abundant outside the cell, where they are a main component of the gel-like extracellular matrix that helps to bind collections of cells together to form tissues. Cells secrete extracellular matrix proteins into their surroundings, where they often assemble into sheets or long fibrils. Collagen is the most abundant of these proteins in animal tissues. A collagen molecule consists of three long polypeptide chains, each containing the nonpolar amino acid glycine at every third position. This regular structure allows the chains to wind around one another to generate a long, regular triple helix (Figure 3–24). Many collagen molecules then bind to one another side-by-side and end-to-end to create long overlapping arrays—thereby generating the extremely tough collagen fibrils that give connective tissues their tensile strength, as described in Chapter 19.
Figure 3–24The fibrous protein collagen. The collagen molecule is a triple helix formed by three extended protein chains that wrap around one another (bottom). In the extracellular space, many rodlike collagen molecules become covalently linked together through their lysine side chains to form collagen fibrils (top) that have the tensile strength of steel. The striping on the collagen fibril is caused by the regular repeating arrangement of the collagen molecules within the fibril.
Covalent Cross-Linkages Stabilize Extracellular Proteins
Many protein molecules are either attached to the outside of a cell’s plasma membrane or secreted to form part of the extracellular matrix. All such proteins are directly exposed to extracellular conditions. To help maintain their structures, the polypeptide chains in such proteins are often stabilized by covalent cross-linkages. These linkages can either tie together two amino acids in the same protein or join together many polypeptide chains in a large protein complex—as for the collagen fibrils just described.
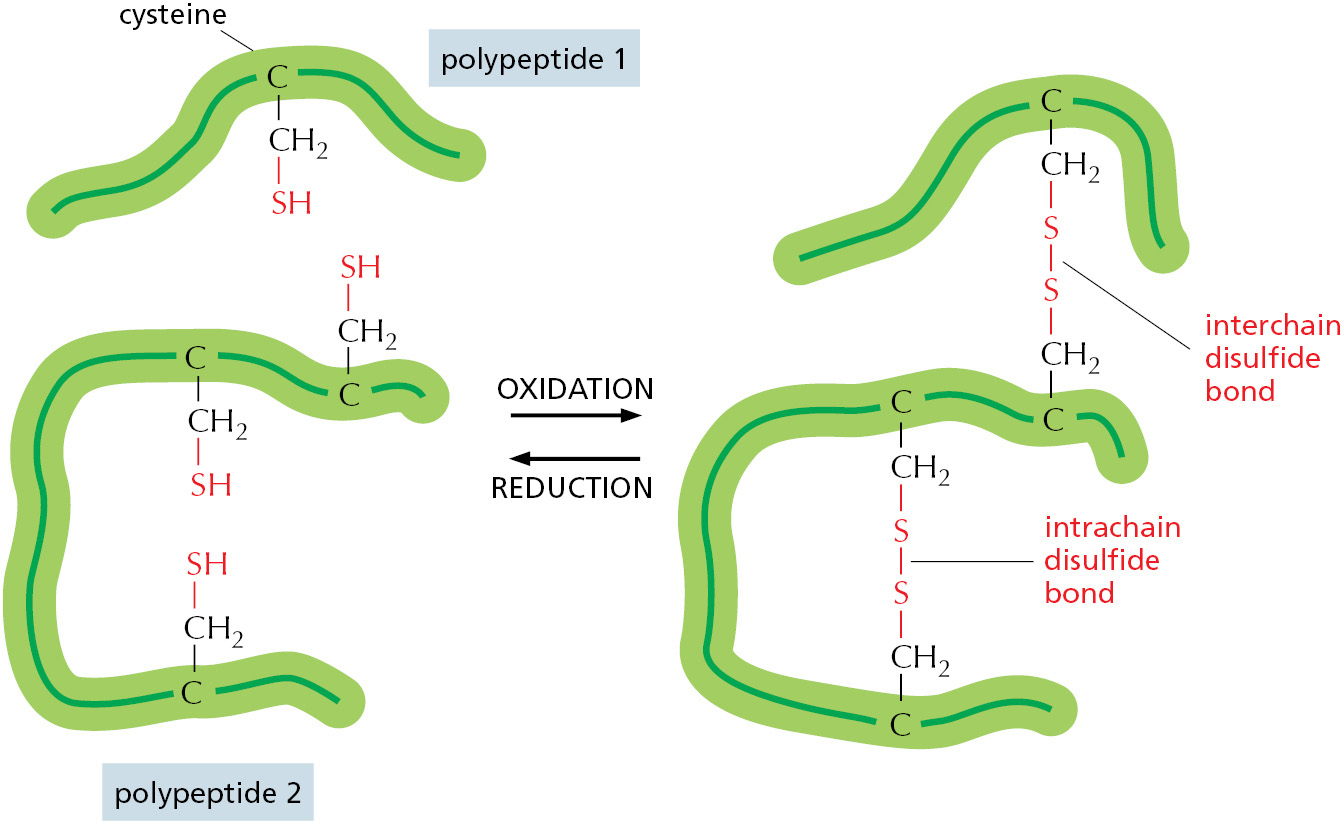
A variety of such cross-links exist, but the most common are covalent sulfur–sulfur bonds. These disulfide bonds (also called S–S bonds) form as cells prepare newly synthesized proteins for export. As described in Chapter 12, their formation is catalyzed in the endoplasmic reticulum by an enzyme that links together the –SH groups of two cysteine side chains that are adjacent in the folded protein (Figure 3–25). Disulfide bonds do not change the conformation of a protein but instead act as atomic staples to reinforce its most favored conformation. For example, lysozyme—an enzyme in tears that dissolves bacterial cell walls—retains its antibacterial activity for a long time because it is stabilized by such cross-linkages.
Figure 3–25Disulfide bonds. Covalent disulfide bonds form between adjacent cysteine side chains. These cross-linkages can join either two parts of the same polypeptide chain or two different polypeptide chains. Because the energy required to break one covalent bond is much larger than the energy required to break even a whole set of noncovalent bonds (see Table 2–1, p. 51), a disulfide bond can have a major stabilizing effect on a protein (Movie 3.7).
Disulfide bonds generally fail to form in the cytosol, where a high concentration of reducing agents converts S–S bonds back to cysteine –SH groups. Apparently, proteins do not require this type of reinforcement in the relatively mild environment inside the cell.
Protein Molecules Often Serve as Subunits for the Assembly of Large Structures
The same principles that enable a protein molecule to associate with itself to form rings or a long filament also operate to generate structures that are formed from a set of different macromolecules, such as enzyme complexes, ribosomes, viruses, and membranes. These much larger objects are not made as single, giant, covalently linked molecules. Instead they are formed by the noncovalent assembly of many separately manufactured molecules, which serve as the subunits of the final structure.
The use of smaller subunits to build larger structures has several advantages:
A large structure built from one or a few repeating smaller subunits requires only a small amount of genetic information.
Both assembly and disassembly can be readily controlled reversible processes, because the subunits associate through multiple bonds of relatively low energy.
Errors in the synthesis of the structure can be more easily avoided, because correction mechanisms can operate during the course of assembly to exclude malformed subunits.
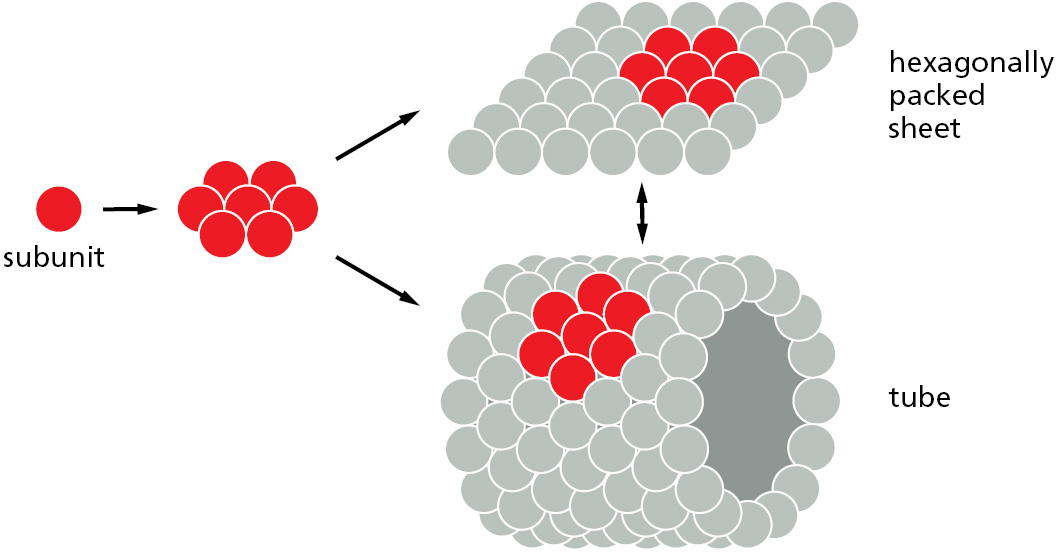
To focus on a well-studied example, we can consider how a virus forms from a mixture of proteins and nucleic acids. Some protein subunits are found to assemble into flat sheets in which the subunits are arranged in hexagonal patterns, but with a slight change in the geometry of the individual subunits, a hexagonal sheet can be converted into a tube (Figure 3–26) or, with more changes, into a hollow sphere. Protein tubes and spheres that bind specific RNA and DNA molecules in their interior form the coats of viruses.
Figure 3–26Single protein subunits form protein assemblies that feature multiple protein–protein contacts. Hexagonally packed globular protein subunits are shown here forming either flat sheets or tubes. Such large structures are not considered to be single “molecules.” Instead, like the actin filament described previously, they are viewed as assemblies formed of many different molecules.
The formation of closed structures, such as rings, tubes, or spheres, provides additional stability because it increases the number of noncovalent bonds between the protein subunits. Moreover, because such a structure is created by mutually dependent, cooperative interactions between subunits, a relatively small change that affects each subunit individually can cause the structure to assemble or disassemble. These principles are dramatically illustrated in the protein coat, or capsid, of many simple viruses, which takes the form of a hollow sphere based on an icosahedron (Figure 3–27). Capsids are often made of hundreds of identical protein subunits that enclose and protect the viral nucleic acid (Figure 3–28). The protein in such a capsid must have a particularly adaptable structure: not only must it make several different kinds of contacts to create the sphere, it must also change this arrangement to let the nucleic acid out to initiate viral replication once the virus has entered a cell.
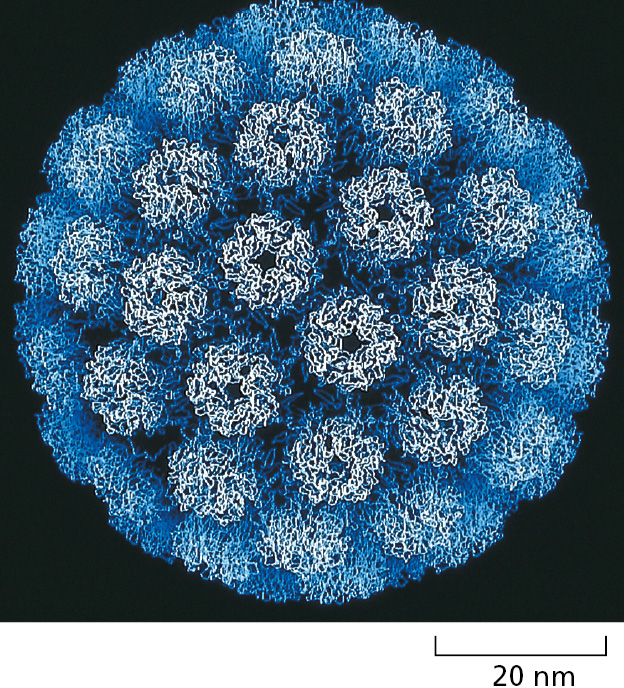
Figure 3–27The protein capsid of a virus. The structure of the simian virus SV40 capsid has been determined by x-ray crystallography and, as for the capsids of many other viruses, it is known in atomic detail. (Courtesy of Robert Grant, Stephan Crainic, and James M. Hogle.)Figure 3–28The structure of a spherical virus. In viruses, many copies of a single protein subunit often pack together to create a spherical shell (a capsid). This capsid encloses the viral genome, composed of either RNA or DNA. For geometric reasons, no more than 60 identical subunits can pack together in a precisely symmetrical way. If slight irregularities are allowed, however, more subunits can be used to produce a larger capsid that retains icosahedral symmetry. The tomato bushy stunt virus (TBSV) shown here, for example, is a spherical virus about 33 nm in diameter formed from 180 identical copies of a 386-amino-acid capsid protein (90 dimers) plus an RNA genome of 4500 nucleotides. To construct such a large capsid, the protein must be able to fit into three somewhat different environments. This requires three slightly different conformations, each of which is differently colored in the virus particle shown here. The postulated pathway of assembly is shown; the precise three-dimensional structure has been determined by x-ray diffraction. (Courtesy of Steve Harrison.)
Many Structures in Cells Are Capable of Self-Assembly
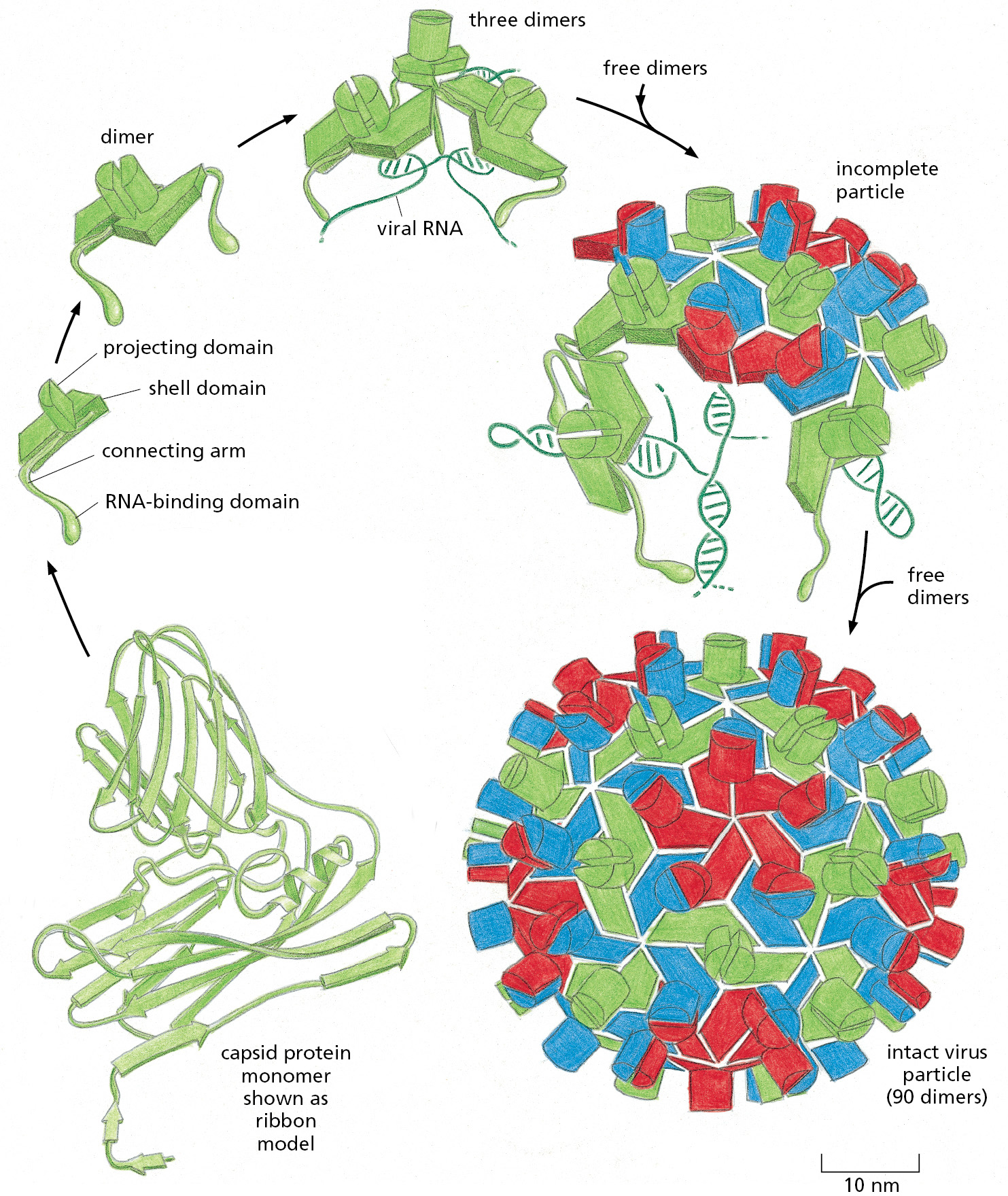
The information for forming many of the complex assemblies of macromolecules in cells must be contained in the subunits themselves, because purified subunits can spontaneously assemble into the final structure under the appropriate conditions. The first large macromolecular aggregate shown to be capable of self-assembly from its component parts was tobacco mosaic virus (TMV). This virus is a long rod in which a cylinder of protein is arranged around a helical RNA core, which constitutes the viral genome (Figure 3–29). If the dissociated RNA and protein subunits are mixed together in solution, they recombine to form fully active viral particles. The assembly process is unexpectedly complex and includes the formation of double rings of protein, which serve as intermediates that add to the growing viral coat.
Figure 3–29The structure of tobacco mosaic virus (TMV). (A) An electron micrograph of the viral particle, which consists of a single long RNA molecule enclosed in a cylindrical protein coat composed of identical protein subunits. (B) A model showing part of the structure of TMV. An RNA molecule of 6395 nucleotides, present as a single strand, is packaged in a helical coat constructed from 2130 copies of a coat protein 158 amino acids long. Fully infective viral particles can self-assemble in a test tube from purified RNA and protein molecules. (A, courtesy of Robley Williams; B, courtesy of Richard J. Feldmann.)
Another complex macromolecular aggregate that can reassemble from its component parts is the bacterial ribosome. This structure is composed of about 55 different protein molecules and 3 different ribosomal RNA (rRNA) molecules. Incubating a mixture of the individual components under appropriate conditions in a test tube causes them to spontaneously re-form the original structure. Most important, such reconstituted ribosomes are able to catalyze protein synthesis. As might be expected, the reassembly of ribosomes follows a specific pathway: after certain proteins have bound to the RNA, this complex is then recognized by other proteins, and so on, until the structure is complete.
It is still not clear how some of the more elaborate self-assembly processes are regulated. Many structures in the cell, for example, have a precisely defined length that appears to be many times greater than that of their component macromolecules. How such length determination is achieved is in many cases a mystery. In the simplest case, a long core protein or other macromolecule provides a scaffold that determines the extent of the final assembly. This is the mechanism that determines the length of the TMV particle, where the RNA chain provides the core. Similarly, a core protein interacting with actin is thought to determine the length of the thin filaments in muscle.
Assembly Factors Often Aid the Formation of Complex Biological Structures
Not all cellular structures held together by noncovalent bonds self-assemble. A cilium, or a myofibril of a muscle cell, for example, cannot form spontaneously from a solution of its component macromolecules. In these cases, part of the assembly information is provided by special enzymes and other proteins that perform the function of templates, serving as assembly factors that guide construction but take no part in the final assembled structure.
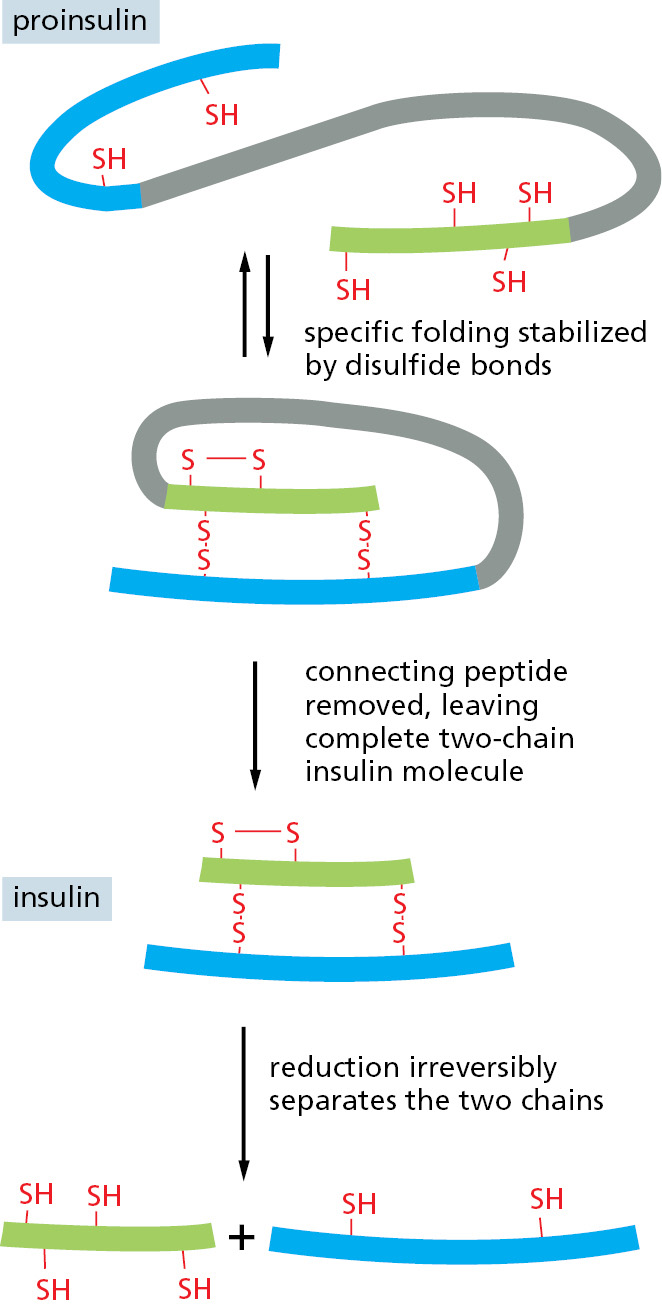
Even relatively simple structures may lack some of the ingredients necessary for their own assembly. In the formation of certain bacterial viruses, for example, the head, which is composed of many copies of a single protein subunit, is assembled on a temporary scaffold composed of a second protein that is produced by the virus. Because the second protein is absent from the final viral particle, the head structure cannot spontaneously reassemble once it has been taken apart. Other examples are known in which proteolytic cleavage is an essential and irreversible step in the normal assembly process. This is even the case for some small protein assemblies, including the structural protein collagen and the hormone insulin (Figure 3–30). From these relatively simple examples, it seems certain that the assembly of a structure as complex as a cilium will involve a temporal and spatial ordering that is imparted by numerous other components.
Figure 3–30Proteolytic cleavage in insulin assembly. The polypeptide hormone insulin cannot spontaneously re-form efficiently if its disulfide bonds are disrupted. It is synthesized as a larger protein (proinsulin) that is cleaved by a proteolytic enzyme after the protein chain has folded into a specific shape. Excision of part of the proinsulin polypeptide chain removes some of the information needed for the protein to fold spontaneously into its normal conformation. For this reason, once insulin has been denatured and its two polypeptide chains have separated, its ability to reassemble is lost.
When Assembly Processes Go Wrong: The Case of Amyloid Fibrils
A special class of protein structure, utilized for some normal cell functions, can also contribute to human diseases when not controlled. These are self-propagating, very stable β-sheet aggregates called amyloid fibrils. These fibrils are built from a series of identical polypeptide chains that become layered one over the other to create a continuous stack of β strands, with each of the β strands oriented perpendicular to a fibril axis (Figure 3–31). In a fibril, two of these stacks of β strands are paired with each other to form a long cross-beta filament, with many hundreds of monomers producing an unbranched fibrous structure that can be several micrometers long and 5–15 nm in width (Figure 3–32). A surprisingly large fraction of proteins have the potential to adopt such structures, because only a short segment of the polypeptide chain is needed to form the spine of the fibril; in addition, the spine can accommodate a variety of amino acid sequences. Nevertheless, very few proteins will actually form this structure inside cells.
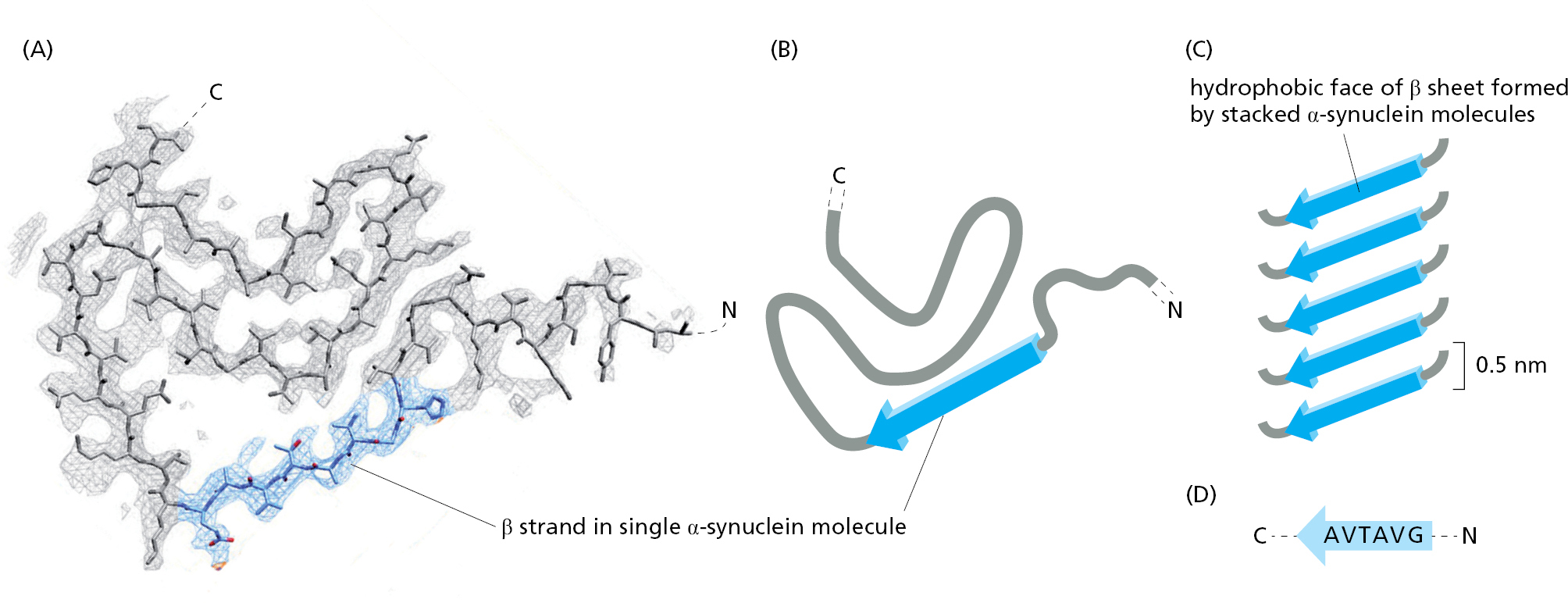
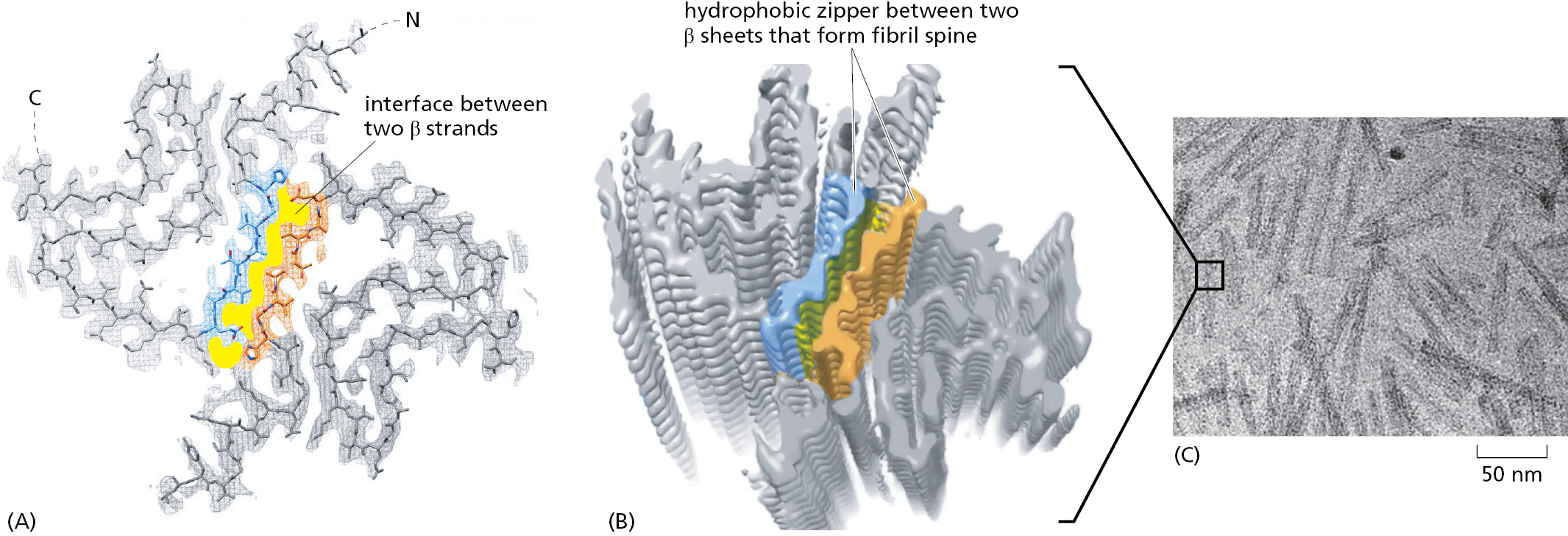
Figure 3–31How an amyloid fibril forms from a protein associated with Parkinson’s disease. Illustrated here is the structure of one-half of an amyloid fibril that is formed by the protein α-synuclein, whose abnormal aggregates contribute to Parkinson’s disease. The conformation of the α-synuclein monomer is shown as an atomic model in (A) and schematically in (B), with the β strand that will form the cross-beta spine of the filament colored blue (only 57 of α-synuclein’s 140 amino acids are shown). (C) How the monomer associates to form a long sheet of stacked β strands. As illustrated in Figure 3–32, a second, identical sheet of β strands pairs with this one to form a two-sheet motif that runs the entire length of the fibril. (D) The amino acid sequence that creates a hydrophobic zipper joining the two sheets, forming the cross-beta spine of the fibril. (From R. Guerrero-Ferreira et al., eLife 7:e36402, 2018.)Figure 3–32The structure of an amyloid fibril. (A) How two monomers of α-synuclein pair to create an amyloid fibril. (B) A three-dimensional rendering of a section of the complete fibril, as determined by cryo-electron microscopy. (C) Electron micrograph of α-synuclein amyloid fibrils. The α-synuclein protein, like some other amyloid-forming proteins, can form several different variants of amyloid fibrils from the same polypeptide chain—only one of which is illustrated here. (From R. Guerrero-Ferreira et al., eLife 7:e36402, 2018. This article is distributed under a Creative Commons Attribution 4.0 International license.)
In humans, the quality-control mechanisms governing proteins gradually decline with age, occasionally permitting normal proteins to form pathological aggregates. In extreme cases, the accumulation of such amyloid fibrils in the cell interior can kill the cells and damage tissues. Because the brain is composed of a highly organized collection of nerve cells that cannot regenerate, the brain is especially vulnerable to this sort of cumulative damage. Thus, although amyloid fibrils may form in different tissues and are known to cause pathologies in several sites in the body, the most severe amyloid pathologies are neurodegenerative diseases. For example, an abnormal formation of amyloid fibrils is thought to play a central causative role in both Alzheimer’s and Parkinson’s diseases.
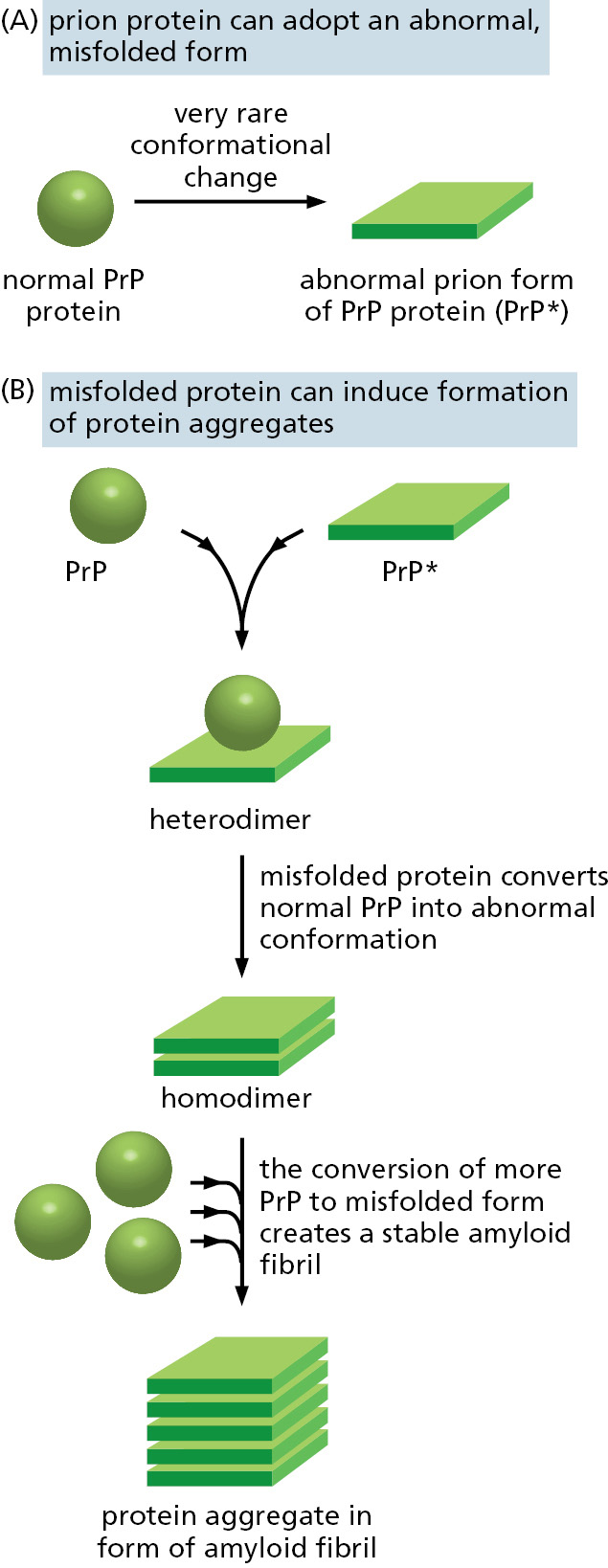
Prion diseases are a special type of these pathologies. They have attained special notoriety because, unlike Parkinson’s or Alzheimer’s, prion diseases can readily spread from one organism to another, providing that the second organism eats a tissue containing the protein aggregate. A set of closely related diseases—scrapie in sheep, Creutzfeldt–Jakob disease (CJD) in humans, kuru in humans, and bovine spongiform encephalopathy (BSE) in cattle—are caused by a misfolded, aggregated form of a particular protein called PrP (for prion protein). PrP is normally located on the outer surface of the plasma membrane, most prominently in neurons, and it has the unfortunate property of forming amyloid fibrils that are “infectious” because they convert normally folded molecules of PrP to the same pathological form (Figure 3–33). This property creates a positive feedback loop that propagates the abnormal form of PrP, called PrP*, and allows the pathological conformation to spread rapidly from cell to cell in the brain, eventually causing death. It can be dangerous to eat the tissues of animals that contain PrP*, as witnessed by the spread of BSE (commonly referred to as “mad cow disease”) from cattle to humans. Fortunately, in the absence of PrP*, PrP is extraordinarily difficult to convert to its abnormal form.
Figure 3–33Prion diseases are caused by proteins whose misfolding is infectious. (A) Schematic illustration of the type of conformational change in the prion protein (PrP) that produces material for an amyloid fibril. (B) The self-infectious nature of the protein aggregation that is central to prion diseases. The misfolded version of the protein, called PrP*, induces the normal PrP protein it contacts to change its conformation, as shown. PrP* is extremely stable, and if eaten, it can produce amyloid fibrils that disrupt brain-cell function, causing a deadly neurodegenerative disorder. Some of the abnormal amyloid fibrils that form in common noninfectious neurodegenerative disorders, including Parkinson’s and Alzheimer’s diseases, appear to propagate from cell to cell within the brain in a similar way.
A closely related protein-only inheritance has been observed in yeast cells. The ability to study infectious proteins in yeast has clarified another remarkable feature of prions. These protein molecules can form several distinctively different types of amyloid fibrils from the same polypeptide chain. Moreover, each type of aggregate can be infectious, forcing normal protein molecules to adopt the same type of abnormal structure. Thus, several different “strains” of infectious particles can arise from the same polypeptide chain.
Recent data suggest that at least some of the abnormal amyloids that form in common human neurological diseases promote the disease by spreading from cell to cell in the brain in a “prion-like” manner, with the abnormally folded form of the protein being taken up by neighboring cells to seed a more widespread formation of the same abnormal structures (for example, α-synuclein in Parkinson’s disease, tau protein in Alzheimer’s disease). Drugs and antibody treatments are currently being designed in attempts to block these spreading events—and thereby reduce the terrible human toll created by these widespread, common diseases.
Amyloid Structures Can Also Perform Useful Functions in Cells
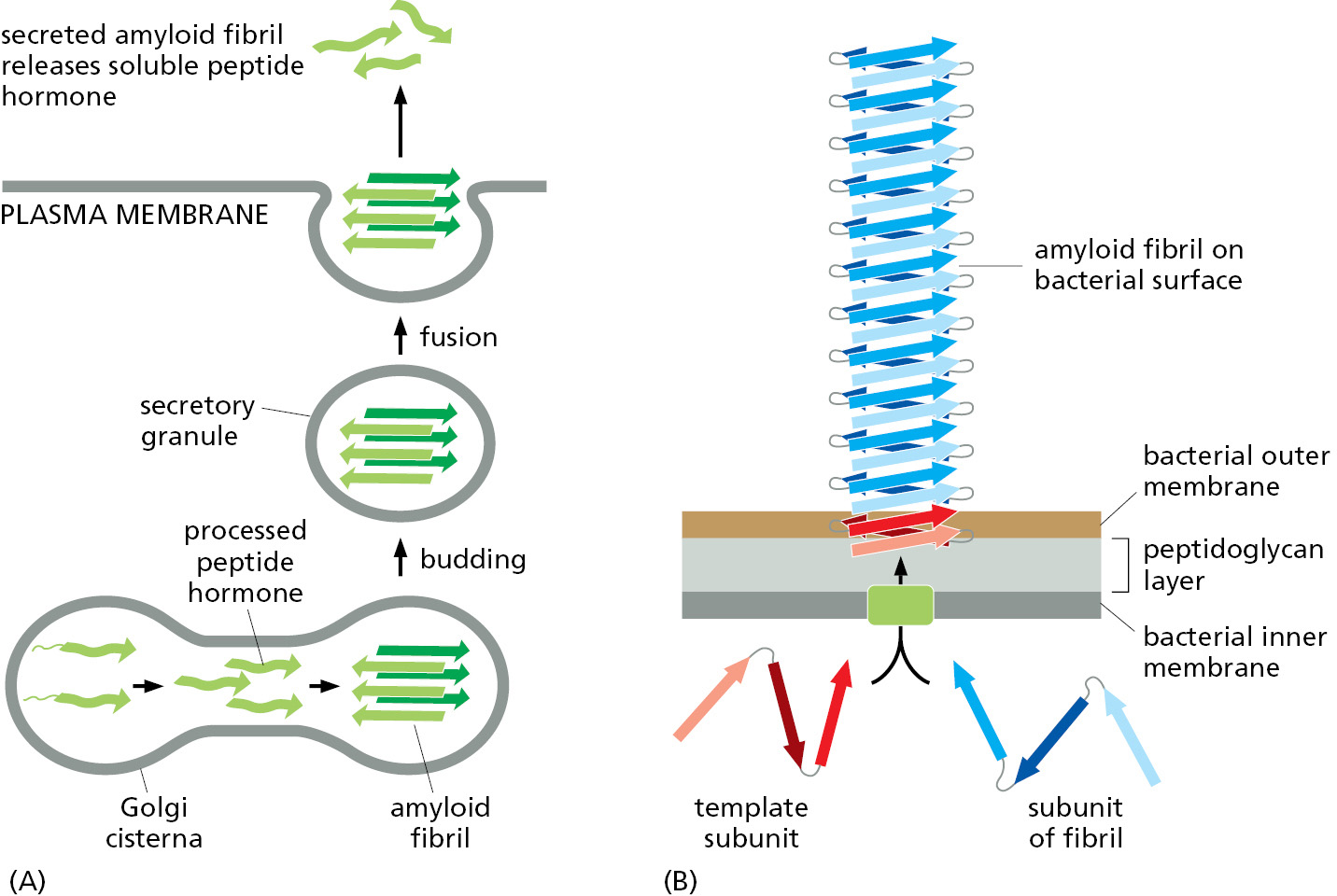
Amyloid fibrils were initially studied because they cause disease. But the same type of structure is now known to be exploited by cells for useful functions. Eukaryotic cells, for example, store many different peptide and protein hormones that they will secrete in specialized secretory vesicles, which package a high concentration of their cargo in dense cores with a regular structure (see Figure 13–43). We now know that these structured cores consist of amyloid fibrils, which in this case have a structure that causes them to dissolve to release soluble cargo after being secreted by exocytosis to the cell exterior (Figure 3–34A). Many bacteria use the amyloid structure in a very different way, secreting proteins that form long amyloid fibrils that project from the cell exterior to help bind bacterial neighbors into biofilms (Figure 3–34B). Because these biofilms help bacteria to survive in adverse environments (including in humans treated with antibiotics), new drugs that specifically disrupt the fibrous networks formed by bacterial amyloids have promise for treating human infections.
Figure 3–34Two normal functions for amyloid fibrils. (A) In eukaryotic cells, protein cargo can be packed very densely in secretory vesicles and stored until signals cause a release of this cargo by exocytosis. For example, proteins and peptide hormones of the endocrine system, such as glucagon and calcitonin, are efficiently stored as short amyloid fibrils, which dissociate when they reach the cell exterior. (B) Bacteria produce amyloid fibrils on their surface by secreting their precursor proteins; these fibrils then create biofilms that link together, and help to protect, large numbers of individual bacteria.
Summary
A protein molecule’s amino acid sequence determines its three-dimensional conformation. Large numbers of noncovalent attractions between different parts of the polypeptide chain stabilize its folded structure. For example, amino acids with hydrophobic side chains tend to cluster in the interior of the molecule, and local hydrogen-bond interactions between neighboring peptide bonds give rise to α helices and β sheets.
Regions of contiguous amino acid sequence fold into globular protein domains. These domains generally contain 40–350 amino acids, and they are the modular units from which larger proteins are constructed. Small proteins typically consist of only a single domain, while large proteins are formed from multiple domains linked together by various lengths of relatively disordered polypeptide chain. As organisms have evolved, the DNA sequences that encode these domains have duplicated, mutated, and been combined with other domains to construct large numbers of new proteins.
Proteins are brought together into larger structures by the same noncovalent attractions that determine protein folding. Proteins with binding sites for their own surface can assemble into dimers, closed rings, spherical shells, or helical polymers. The amyloid fibril is a long unbranched structure assembled through a repeating aggregate of β sheets.
Some mixtures of proteins and nucleic acids can assemble spontaneously into complex structures in a test tube. But not all structures in the cell are capable of spontaneous reassembly after they have been dissociated into their component parts, because many biological assembly processes involve assembly factors that have been removed from the final structure.
The part of an amino acid that differs between amino acid types. The side chains give each type of amino acid its unique physical and chemical properties.
Common folding pattern in proteins, in which a linear sequence of amino acids folds into a right-handed helix stabilized by internal hydrogen-bonding between backbone atoms.
Common structural motif in proteins in which different sections of the polypeptide chain run alongside each other, joined together by hydrogen-bonding between atoms of the polypeptide backbone. Also known as a β pleated sheet.
(protein domain) Portion of a protein that has a tertiary structure of its own. Larger proteins are generally composed of several domains, each connected to the next by short flexible regions of polypeptide chain. Homologous domains are recognized in many different proteins.
Self-propagating, stable β-sheet aggregates built from hundreds of identical polypeptide chains that become layered one over the other to create a continuous stack of β sheets. The unbranched fibrous structure can contribute to human diseases when not controlled.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}