We have seen that each type of protein consists of a precise sequence of amino acids that allows it to fold up into a particular three-dimensional shape, or conformation. These proteins can also have moving parts whose mechanical actions are coupled to chemical events. This coupling of chemistry and movement helps to give proteins the extraordinary capabilities that underlie the dynamic processes in living cells.
In this section, we explain how proteins bind to other selected molecules and how a protein’s activity depends on such binding. We will use selected examples to demonstrate how their ability to bind to other molecules enables proteins to act as catalysts, signal receptors, switches, motors, or tiny pumps. These examples by no means exhaust the vast functional repertoire of proteins. You will encounter the specialized functions of many other proteins elsewhere in this book, based on similar principles.
All Proteins Bind to Other Molecules
A protein molecule’s physical interaction with other molecules determines its biological properties. Thus, antibodies attach to viruses or bacteria to mark them for destruction, the enzyme hexokinase binds glucose and ATP so as to catalyze a reaction between them, actin molecules bind to each other to assemble into actin filaments, and so on. Indeed, all proteins stick, or bind, to other molecules. In some cases, this binding is very tight; in others it is weak and short-lived. But the binding always shows great specificity, in the sense that each protein molecule can usually bind just one or a few molecules out of the many thousands of different types it encounters. The substance that is bound by the protein—whether it is an ion, a small molecule, or a macromolecule such as another protein—is referred to as a ligand for that protein (from the Latin word ligare, meaning “to bind”).
The ability of a protein to bind selectively and with high affinity to a ligand depends on the formation of a set of weak noncovalent bonds—hydrogen bonds, electrostatic attractions, and van der Waals attractions—plus favorable hydrophobic interactions (see Panel 2–3, pp. 98–99). Because each individual bond is weak, effective binding occurs only when many of these bonds form simultaneously. Such binding is possible only if the surface contours of the ligand molecule fit very closely to the protein, matching it like a hand in a glove (Figure 3–35).
Figure 3–35The selective binding of a protein to another molecule. Many weak bonds are needed to enable a protein to bind tightly to a second molecule, or ligand. A ligand must therefore fit precisely into a protein’s binding site, like a hand into a glove, so that a large number of noncovalent bonds form between the protein and the ligand. (A) Schematic; (B) space-filling model. (PDB code: 1G6N.)
The region of a protein that associates with a ligand, known as the ligand’s binding site, usually consists of a cavity in the protein surface formed by a particular arrangement of amino acids. These amino acids can belong to different portions of the polypeptide chain that are brought together when the protein folds (Figure 3–36). Separate regions of the protein surface generally provide binding sites for different ligands, allowing the protein’s activity to be regulated, as we shall see later. And other parts of the protein act as a handle to position the protein in the cell—an example is the SH2 domain discussed previously, which often moves a protein containing it to particular intracellular sites in response to signals.
Figure 3–36The binding site of a protein. (A) The folding of the polypeptide chain typically creates a crevice or cavity on the protein surface. This crevice contains a set of amino acid side chains disposed in such a way that they can form noncovalent bonds only with certain ligands. (B) A close-up of an actual binding site, showing the hydrogen bonds and electrostatic interactions formed between a protein and its ligand. In this example, a molecule of cyclic AMP is the bound ligand, shown in dark yellow.
Although the atoms buried in the interior of the protein have no direct contact with the ligand, they form the framework that gives the surface its contours and its chemical and mechanical properties. Even small changes to the amino acids in the interior of a protein molecule can change its three-dimensional shape enough to destroy a binding site on the surface.
The Surface Conformation of a Protein Determines Its Chemistry
The impressive chemical capabilities of proteins often require that the chemical groups on their surface interact in ways that enhance the chemical reactivity of one or more amino acid side chains. These interactions fall into two main categories.
First, the interaction of neighboring parts of the polypeptide chain may restrict the access of water molecules to that protein’s ligand-binding sites. Because water molecules readily form hydrogen bonds that can compete with ligands for sites on the protein surface, a ligand will form tighter hydrogen bonds (and electrostatic interactions) with a protein if water molecules are kept away. It might be hard to imagine a mechanism that would exclude a molecule as small as water from a protein surface without affecting the access of the ligand itself. However, because of the strong tendency of water molecules to form water–water hydrogen bonds, water molecules exist in a large hydrogen-bonded network (see Panel 2–2, pp. 96–97). In effect, a protein can keep a ligand-binding site dry, increasing that site’s reactivity, because it is energetically unfavorable for individual water molecules to break away from this network—as they must do to reach into a crevice on a protein’s surface.
Second, the clustering of neighboring polar amino acid side chains can alter their reactivity. If protein folding brings together a number of negatively charged side chains against their mutual repulsion, for example, the affinity of the site for a positively charged ion is greatly increased. In addition, when amino acid side chains interact with one another through hydrogen bonds, normally unreactive groups (such as the –CH2OH on the serine shown in Figure 3–37) can become reactive, enabling them to be used to make or break selected covalent bonds.
Figure 3–37An unusually reactive amino acid at the active site of an enzyme. This example is the catalytic triad Asp-His-Ser found in chymotrypsin, elastase, and other serine proteases (see Figure 3–13). The aspartic acid side chain (Asp) induces the histidine (His) to remove the proton from a particular serine (Ser). This activates the serine and enables it to form a covalent bond with the enzyme’s substrate, hydrolyzing a peptide bond. The many convolutions of the polypeptide chain are omitted here.
The surface of each protein molecule therefore has a unique chemical reactivity that depends not only on which amino acid side chains are exposed, but also on their exact orientation relative to one another. For this reason, two slightly different conformations of the same protein molecule can differ greatly in their chemistry.
Sequence Comparisons Between Protein Family Members Highlight Crucial Ligand-binding Sites
As we have described previously, genome sequences allow us to group many of the domains found in proteins into families that show clear evidence of their evolution from a common ancestor. The three-dimensional structures of members of the same domain family are remarkably similar. For example, even when the amino acid sequence identity falls to 25%, the backbone atoms in a domain can follow a common protein fold within 0.2 nm (2 Å).
We can use a method called evolutionary tracing to identify those sites in a protein domain that are the most crucial to the domain’s function. Those sites that bind to other molecules are the most likely to be kept unchanged as organisms evolve. Thus, in this method, those amino acids that are the same, or nearly so, in all of the known protein family members are mapped onto a model of the three-dimensional structure of a single family member. When this is done, the most invariant positions often form one or more clusters on the protein surface, as illustrated in Figure 3–38A for the SH2 domain described previously (see Figure 3–9). These clusters generally correspond to ligand-binding sites.
The SH2 domain functions to link two proteins together. It binds the protein containing it to a second protein that contains a phosphorylated tyrosine side chain in a specific amino acid sequence context, as shown in Figure 3–38B. The amino acids located at the binding site for the phosphorylated polypeptide have been the slowest to change during the long evolutionary process that produced the large SH2 family of peptide recognition domains. Mutation is a random process; survival is not. Thus, natural selection (random mutation followed by nonrandom survival) produces the sequence conservation by preferentially eliminating organisms whose SH2 domains have become altered in a way that inactivates the SH2 binding site, destroying SH2 function.
Figure 3–38The evolutionary trace method applied to a protein domain. (A) Front and back views of a space-filling model of the SH2 domain, with evolutionarily conserved amino acids on the protein surface colored yellow, and those more toward the protein interior colored red. (B) The structure of one specific SH2 domain with its bound polypeptide. Here, those amino acids located within 0.4 nm of the bound ligand are colored blue. The two key amino acids of the ligand are yellow, and the others are purple. Note the high degree of correspondence between A and B. (Adapted from O. Lichtarge et al., J. Mol. Biol. 257:342–358, 1996. PDB codes: 1SPR, 1SPS.)
Genome sequencing has revealed huge numbers of proteins whose functions are unknown. Once a three-dimensional structure has been determined for one member of a protein family, evolutionary tracing allows biologists to determine binding sites for the members of that family, and this provides a useful start in deciphering protein function.
Proteins Bind to Other Proteins Through Several Types of Interfaces
Proteins can bind to other proteins in multiple ways. In many cases, a portion of the surface of one protein contacts an extended loop of polypeptide chain (a string) on a second protein (Figure 3–39A). Such a surface–string interaction, for example, allows the SH2 domain to recognize a phosphorylated polypeptide loop on a second protein, as just described, and it also enables a protein kinase to recognize the proteins that it will phosphorylate (see below).
Figure 3–39Three ways in which two proteins can bind to each other. Only the interacting parts of the two proteins are shown. (A) A rigid surface on one protein can bind to an extended loop of polypeptide chain (a string) on a second protein. (B) Two α helices can bind together to form a coiled-coil. (C) Two complementary rigid surfaces often link two proteins together. Binding interactions can also involve the pairing of β strands (see, for example, Figure 3–19B).
A second type of protein–protein interface forms when two α helices, one from each protein, pair together to form a coiled-coil (Figure 3–39B). This type of protein interface is found in several families of transcription regulatory proteins, as discussed in Chapter 7.
Another common way for proteins to interact is by the precise matching of one rigid surface with that of another (Figure 3–39C). Such interactions can be very tight, because a large number of weak bonds can form between two surfaces that match well. For the same reason, such surface–surface interactions can be extremely specific, enabling a protein to select just one partner from the many thousands of different proteins found in a cell.
Antibody Binding Sites Are Especially Versatile
All proteins must bind to particular ligands to carry out their various functions, and the antibody family is notable for its capacity for tight, highly selective binding (see Chapter 24).
Antibodies, or immunoglobulins, are proteins produced by the immune system in response to foreign molecules, such as those on the surface of an invading microorganism. Each antibody binds tightly to a particular target molecule, thereby either inactivating the target molecule directly or marking it for destruction. An antibody recognizes its target (called an antigen) with remarkable specificity. Because there are potentially billions of different antigens that humans might encounter, we need to be able to produce billions of different antibodies.
Antibodies are Y-shaped molecules with two identical binding sites that are complementary to a small portion of the surface of the antigen molecule. A detailed examination of the antigen-binding sites of antibodies reveals that they are formed from several loops of polypeptide chain that protrude from the ends of a pair of closely juxtaposed protein domains (Figure 3–40). The genes that encode different antibodies generate an enormous diversity of antigen-binding sites by changing only the length and amino acid sequence of these loops, without altering the basic protein structure.
Figure 3–40An antibody is Y-shaped and has two identical antigen-binding sites, one on each arm of the Y. (A) Schematic drawing of a typical antibody molecule. The protein is composed of four polypeptide chains (two identical heavy chains and two identical, smaller light chains), stabilized and held together by disulfide bonds (red). Each chain is made up of several similar domains, here shaded with blue, for the variable domains, or gray, for the constant domains. The antigen-binding site is formed where a heavy-chain variable domain (VH) and a light-chain variable domain (VL) come close together. These are the domains that differ most in their amino acid sequence in different antibodies—hence their name. (B) Ribbon drawing of a single light chain showing that the most variable parts of the polypeptide chain (orange) extend as loops at one end of the variable domain (VL) to form half of one antigen-binding site of the antibody molecule shown in A. Note that both the constant and variable domains are composed of a sandwich of two antiparallel β sheets connected by a disulfide bond (red). (See Movie 24.5.)
Loops of this kind are ideal for grasping other molecules. They allow a large number of chemical groups to surround a ligand so that the protein can link to it with many weak bonds. For this reason, loops often form the ligand-binding sites in proteins.
The Equilibrium Constant Measures Binding Strength
Molecules in the cell encounter each other very frequently because of their continual random thermal movements. Colliding molecules with poorly matching surfaces form few noncovalent bonds with one another, and the two molecules dissociate as rapidly as they come together. At the other extreme, when many noncovalent bonds form between two colliding molecules, the association can persist for a very long time (Figure 3–41). Such strong interactions occur in cells whenever a biological function requires that molecules remain associated; for example, when a group of RNA and protein molecules come together to make a subcellular structure such as a ribosome.
Figure 3–41How noncovalent bonds mediate interactions between macromolecules (see Movie 2.1).
We can measure the strength with which any two molecules bind to each other. As an example, consider a population of identical antibody molecules that suddenly encounters a population of ligands diffusing in the fluid surrounding them. At frequent intervals, one of the ligand molecules will bump into the binding site of an antibody and form an antibody–ligand complex. The population of antibody–ligand complexes will therefore increase, but not without limit: over time, a second process, in which individual complexes break apart because of thermally induced motion, will become increasingly important. Eventually, any population of antibody molecules and ligands will reach a steady state, or equilibrium, in which the number of binding (association) events per second is precisely equal to the number of “unbinding” (dissociation) events (see Figure 2–30).
From the concentrations of the ligand, antibody, and antibody–ligand complex at equilibrium, we can calculate a convenient measure of the strength of binding—the equilibrium constant (K); (Figure 3–42A). This constant was described in detail in Chapter 2, where its connection to free-energy differences was derived (see pp. 68–69). The equilibrium constant for a reaction in which two molecules (A and B) bind to each other to form a complex (AB) has units of liters/mole, and half of the binding sites will be occupied by ligand when that ligand’s concentration (in moles/liter) reaches a value that is equal to 1/K. This equilibrium constant is larger the greater the binding strength, and it is a direct measure of the free-energy difference between the bound and free states (Figure 3–42B). Even a change of a few noncovalent bonds can have a striking effect on a binding interaction, as shown by the example in Figure 3–43. (Note that the equilibrium constant, as defined here, is the association or affinity constant, Ka; the reciprocal of Ka is the dissociation constant, Kd, which is also widely used.)
Figure 3–42Relating standard free-energy difference (ΔG°) to the equilibrium constant (K). (A) The equilibrium between molecules A and B and the complex AB is maintained by a balance between the two opposing reactions shown in panels 1 and 2. Molecules A and B must collide if they are to react, and the association rate is therefore proportional to the product of their individual concentrations [A] × [B]. (Square brackets indicate concentration.) As shown in panel 3, the ratio of the rate constants for the association and the dissociation reactions is equal to the equilibrium constant (K) for the reaction. (B) The equilibrium constant in panel 3 is that for the reaction A + B ⇌ AB, and the larger its value, the stronger the binding between A and B. Note that for every 5.91 kJ/mole decrease in standard free energy, the equilibrium constant increases by a factor of 10 at 37°C.
The equilibrium constant here has units of liters/mole; for simple binding interactions it is also called the affinity constant or association constant, denoted Ka. The reciprocal of Ka is called the dissociation constant, Kd (in units of moles/liter).
Figure 3–43Small changes in the number of weak bonds can have drastic effects on a binding interaction. This example illustrates the dramatic effect of the presence or absence of a few weak noncovalent bonds in a biological context.
We have used the case of an antibody binding to its ligand to illustrate the effect of binding strength on the equilibrium state, but the same principles apply to any molecule and its ligand. Many proteins are enzymes, which, as we now discuss, first bind to their ligands and then catalyze the breakage or formation of covalent bonds in these molecules.
Enzymes Are Powerful and Highly Specific Catalysts
Many proteins can perform their function simply by binding to another molecule. An actin molecule, for example, need only associate with other actin molecules to form a filament. There are other proteins, however, for which ligand binding is only a necessary first step in their function. This is the case for the large and very important class of proteins called enzymes. As described in Chapter 2, enzymes are remarkable molecules that cause the chemical transformations that make and break covalent bonds in cells. They bind to one or more ligands, called substrates, and convert them into one or more chemically modified products, doing this over and over again with amazing rapidity. Enzymes speed up reactions, often by a factor of a million or more, without themselves being changed; that is, they act as catalysts that permit cells to make or break covalent bonds in a controlled way. It is the catalysis of organized sets of chemical reactions by enzymes that creates and maintains the cell, making life possible.
We can group enzymes into functional classes that perform similar chemical reactions (Table 3–1). Each type of enzyme within such a class is highly specific, catalyzing only a single type of reaction. Thus, hexokinase adds a phosphate group to D-glucose but ignores its optical isomer L-glucose; the blood-clotting enzyme thrombin cuts one type of blood protein between a particular arginine and its adjacent glycine and nowhere else, and so on. As discussed in detail in Chapter 2, enzymes work in teams, with the product of one enzyme becoming the substrate for the next. The result is an elaborate network of metabolic pathways that provides the cell with energy and generates the many large and small molecules that the cell needs (see Figure 2–62).
TABLE 3–1 Some Common Types of Enzymes
Enzyme
Reaction catalyzed
Hydrolases
General term for enzymes that catalyze a hydrolytic cleavage reaction; nucleases and proteases are more specific names for subclasses of these enzymes
Nucleases
Break down nucleic acids by hydrolyzing bonds between nucleotides. Endonucleases and exonucleases cleave nucleic acids within and from the ends of the polynucleotide chains, respectively
Proteases
Break down proteins by hydrolyzing bonds between amino acids
Synthases
Synthesize molecules in anabolic reactions by condensing two smaller molecules together
Ligases
Join together (ligate) two molecules in an energy-dependent process. DNA ligase, for example, joins two DNA molecules together end-to-end through phosphodiester bonds
Isomerases
Catalyze the rearrangement of bonds within a single molecule
Polymerases
Catalyze polymerization reactions such as the synthesis of DNA and RNA
Kinases
Catalyze the addition of a phosphate group to a molecule. Protein kinases are an important group of kinases that attach phosphate groups to proteins
Phosphatases
Catalyze the hydrolytic removal of a phosphate group from a molecule
Oxido-reductases
General name for enzymes that catalyze reactions in which one molecule is oxidized while the other is reduced. Enzymes of this type are often more specifically named oxidases, reductases, or dehydrogenases
ATPases
Hydrolyze ATP. Many proteins with a wide range of roles have an energy-harnessing ATPase activity as part of their function; for example, motor proteins such as myosin and membrane transport proteins such as the sodium–potassium pump
GTPases
Hydrolyze GTP. A large family of GTP-binding proteins are GTPases with central roles in the regulation of cell processes
Enzyme names typically end in “-ase,” with the exception of some enzymes, such as pepsin, trypsin, thrombin, and lysozyme, that were discovered and named before the convention became generally accepted at the end of the nineteenth century. The common name of an enzyme usually indicates the substrate or product and the nature of the reaction catalyzed. For example, citrate synthase catalyzes the synthesis of citrate by a reaction between acetyl CoA and oxaloacetate.
Substrate Binding Is the First Step in Enzyme Catalysis
For a protein that catalyzes a chemical reaction (an enzyme), the binding of each substrate molecule to the protein is an essential prelude. In the simplest case, if we denote the enzyme by E, the substrate by S, and the product by P, the basic reaction path is E + S → ES → EP → E + P. As illustrated in Figure 3–44, there is a limit to the amount of substrate that a single enzyme molecule can process in a given time. Although an increase in the concentration of substrate increases the rate at which product is formed, this rate eventually reaches a maximum value. At that point the enzyme molecule is saturated with substrate, and the rate of reaction (Vmax) depends only on how rapidly the enzyme can process the substrate molecule. This maximum rate divided by the enzyme concentration is called the turnover number. Turnover numbers are often about 1000 substrate molecules processed per second per enzyme molecule, although turnover numbers between 1 and 10,000 are known.
Figure 3–44Enzyme kinetics. The rate of an enzyme reaction (V) increases as the substrate concentration increases until a maximum value (Vmax) is reached. At this point all substrate-binding sites on the enzyme molecules are fully occupied, and the rate of reaction is limited by the rate of the catalytic process on the enzyme surface. For most enzymes, the concentration of substrate at which the reaction rate is half-maximal (Km) is a measure of how tightly the substrate is bound, with a large value of Km corresponding to weak binding (Km approximates the dissociation constant, Kd, for substrate binding).
The other kinetic parameter frequently used to characterize an enzyme is its Km, the concentration of substrate that allows the reaction to proceed at one-half its maximum rate (0.5Vmax) (see Figure 3–44). A low Km value means that the enzyme reaches its maximum catalytic rate at a low concentration of substrate and generally indicates that the enzyme binds to its substrate very tightly, whereas a high Km value corresponds to weak binding. The methods used to characterize enzymes in this way are explained in Panel 3–2 (pp. 150–151).
Enzymes Speed Reactions by Selectively Stabilizing Transition States
Figure 3–45Enzymes accelerate chemical reactions by decreasing the activation energy. There is a single transition state in this example. However, often both the uncatalyzed reaction (A) and the enzyme-catalyzed reaction (B) go through a series of transition states. In that case, it is the transition state with the highest energy (ST and EST) that determines the activation energy and limits the rate of the reaction. (S = substrate; P = product of the reaction; ES = enzyme–substrate complex; EP = enzyme–product complex.)
Enzymes achieve extremely high rates of chemical reaction—rates that are far higher than for any synthetic catalysts. There are several reasons for this efficiency. First, when two molecules need to react, the enzyme greatly increases the local concentration of both of these substrate molecules at the catalytic site, holding them in the correct orientation for the reaction that is to follow. More important, however, some of the binding energy contributes directly to the catalysis. Substrate molecules must pass through a series of intermediate states of altered geometry and electron distribution before they form the ultimate products of the reaction. The free energy required to attain the most unstable intermediate state, called the transition state, is known as the activation energy for the reaction, and it is the major determinant of the reaction rate. Enzymes have a much higher affinity for the transition state of the substrate than they have for the stable form. Because this tight binding greatly lowers the energy of the transition state, the enzyme greatly accelerates a particular reaction by lowering the activation energy that is required (Figure 3–45; see also p. 63).
Enzymes Can Use Simultaneous Acid and Base Catalysis
Figure 3–46 compares the spontaneous reaction rates and the corresponding enzyme-catalyzed rates for five enzymes. Rate accelerations range from 109 to 1023. This is possible because enzymes not only bind tightly to a transition state, they also contain precisely positioned atoms that alter the electron distributions in the atoms that participate directly in the making and breaking of covalent bonds. Peptide bonds, for example, can be hydrolyzed in the absence of an enzyme by exposing a polypeptide to either a strong acid or a strong base. Enzymes are unique, however, in being able to use acid and base catalysis simultaneously, because the rigid framework of the protein constrains the acidic and basic residues and prevents them from combining with each other, as they would do in solution (Figure 3–47).
Figure 3–46The rate accelerations caused by five different enzymes. (Adapted from A. Radzicka and R. Wolfenden, Science 267:90–93, 1995.)Figure 3–47Simultaneous acid catalysis and base catalysis by an enzyme. (A) The start of the uncatalyzed reaction that hydrolyzes a peptide bond, with blue shading used to indicate electron distribution in the water and carbonyl bonds. (B) An acid likes to donate a proton (H+) to other atoms. By pairing with the carbonyl oxygen, an acid causes electrons to move away from the carbonyl carbon, making this atom much more attractive to the electronegative oxygen of an attacking water molecule. (C) A base likes to take up H+. By pairing with a hydrogen of the attacking water molecule, a base causes electrons to move toward the water oxygen, making it a better attacking group for the carbonyl carbon. (D) By having appropriately positioned atoms on its surface, an enzyme can perform both acid catalysis and base catalysis at the same time. (E) A tetrahedral intermediate is formed by the attack of the water oxygen atom on the carbonyl carbon atom, and this intermediate rapidly decays to hydrolysis products. The red arrows denote the electron shifts associated with product formation.
The fit between an enzyme and its substrate needs to be precise. A small change introduced by genetic engineering in the active site of an enzyme can therefore have a profound effect. Replacing a glutamic acid with an aspartic acid in one enzyme, for example, shifts the position of the catalytic carboxylate ion by only 1 Å (about the radius of a hydrogen atom), yet this is enough to decrease the activity of the enzyme a thousandfold.
Lysozyme Illustrates How an Enzyme Works
To demonstrate how enzymes catalyze chemical reactions, we examine an enzyme that acts as a natural antibiotic in egg white, saliva, tears, and other secretions. Lysozyme catalyzes the cutting of polysaccharide chains in the cell walls of bacteria. The bacterial cell is under pressure from osmotic forces, and cutting even a small number of these chains causes the cell wall to rupture and the cell to burst. A relatively small and stable protein that can be easily isolated in large quantities, lysozyme was the first enzyme to have its structure worked out in atomic detail by x-ray crystallography (in the mid-1960s).
The reaction that lysozyme catalyzes is a hydrolysis: it adds a molecule of water to a single bond between two adjacent sugar groups in the polysaccharide chain, thereby causing the bond to break (see Figure 2–9). The reaction is energetically favorable because the free energy of the severed polysaccharide chain is lower than the free energy of the intact chain. However, there is an energy barrier for the reaction (its activation energy). In particular, a colliding water molecule can break a bond linking two sugars only if the polysaccharide molecule is distorted into a particular shape—the transition state—in which the atoms around the bond have an altered geometry and electron distribution. Because of this requirement, random collisions must supply a very large activation energy for the reaction to take place. In an aqueous solution at room temperature, the energy of collisions almost never exceeds the activation energy. The pure polysaccharide can therefore remain for years in water without being hydrolyzed to any detectable degree.
PANEL 3–2: Some of the Methods Used to Study Enzymes
WHY ANALYZE THE KINETICS OF ENZYMES?
Enzymes are the most selective and powerful catalysts known. An understanding of their detailed mechanisms provides a critical tool for the discovery of new drugs, for the large-scale industrial synthesis of useful chemicals, and for appreciating the chemistry of cells and organisms. A detailed study of the rates of the chemical reactions that are catalyzed by a purified enzyme—more specifically how these rates change with changes in conditions such as the concentrations of substrates, products, inhibitors, and regulatory ligands—allows biochemists to figure out exactly how each enzyme works. For example, this is the way that the ATP-producing reactions of glycolysis, shown previously in Figure 2-47, were deciphered—allowing us to appreciate the rationale for this critical enzymatic pathway.
In this Panel, we introduce the important field of enzyme kinetics, which has been indispensable for deriving much of the detailed knowledge that we now have about cell chemistry.
STEADY-STATE ENZYME KINETICS
Many enzymes have only one substrate, which they bind and then process to produce products according to the scheme outlined in Figure 3–48A. In this case, the reaction is written as
Here we have assumed that the reverse reaction, in which E + P recombine to form EP and then ES, occurs so rarely that we can ignore it. In this case, EP need not be represented, and we can express the rate of the reaction, known as its velocity, V, as
V = kcat[ES]
where [ES] is the concentration of the enzyme-substrate complex, and kcat is the turnover number, a rate constant that has a value equal to the number of substrate molecules processed per enzyme molecule each second.
But how does the value of [ES] relate to the concentrations that we know directly, which are the total concentration of the enzyme, [Eo], and the concentration of the substrate, [S]? When enzyme and substrate are first mixed, the concentration [ES] will rise rapidly from zero to a so-called steady-state level, as illustrated below.
At this steady state, [ES] is nearly constant, so that
or, because the concentration of the free enzyme, [E], is equal to [Eo] - [ES],
Rearranging, and defining the constant Km as
we get
or, remembering that V = kcat[ES], we obtain the famous Michaelis-Menten equation
As [S] is increased to higher and higher levels, essentially all of the enzyme will be bound to substrate at steady state; at this point, a maximum rate of reaction, Vmax, will be reached where V = Vmax = kcat[Eo]. Thus, it is convenient to rewrite the Michaelis-Menten equation as
THE DOUBLE-RECIPROCAL PLOT
A typical plot of V versus [S] for an enzyme that follows Michaelis–Menten kinetics is shown below. From this plot, neither the value of Vmax nor of Km is immediately clear.
To obtain Vmax and Km from such data, a double-reciprocal plot is often used, in which the Michaelis–Menten equation has merely been rearranged, so that 1/V can be plotted versus 1/[S].
THE SIGNIFICANCE OF Km, kcat, and kcat/Km
As described in the text, Km is an approximate measure of substrate affinity for the enzyme: it is numerically equal to the concentration of [S] at V = 0.5Vmax. In general, a lower value of Km means tighter substrate binding. In fact, for those cases where kcat is much smaller than k–1, the Kmwill be equal to Kd, the dissociation constant for substrate binding to the enzyme (Kd = 1/Ka).
We have seen that kcat is the turnover number for the enzyme. At very low substrate concentrations, where [S] << Km, most of the enzyme is free. Thus we can think of [E] = [Eo], so that the Michaelis-Menten equation can be simplified as V = kcat/Km[E][S]. Thus, the ratio kcat/Km is equivalent to the rate constant for the reaction between free enzyme and free substrate.
A comparison of kcat/Km for the same enzyme with different substrates, or for two enzymes with their different substrates, is widely used as a measure of enzyme effectiveness.
For simplicity, in this Panel we have discussed enzymes that have only one substrate, such as the lysozyme enzyme described in the text (see p. 152). Most enzymes have two substrates, one of which is often an active carrier molecule—such as NADH or ATP.
A similar, but more complex, analysis is used to determine the kinetics of such enzymes—allowing the order of substrate binding and the presence of covalent intermediates along the pathway to be revealed.
SOME ENZYMES ARE DIFFUSION LIMITED
The values of kcat, Km, and kcat/Km for some selected enzymes are given below:
enzyme
substrate
kcat(sec–1)
Km(M)
kcat/Km(sec–1 M–1)
acetylcholinesterase
acetylcholine
1.4 × 104
9 × 10–5
1.6 × 108
catalase
H2O2
4 × 107
1
4 × 107
fumarase
fumarate
8 × 102
5 × 10–6
1.6 × 108
Because an enzyme and its substrate must collide before they can react, kcat/Km has a maximum possible value that is limited by collision rates. If every collision forms an enzyme-substrate complex, one can calculate from diffusion theory that kcat/Km will be between 108 and 109 sec–1 M–1, in the case where all subsequent steps proceed immediately. Thus, it is claimed that enzymes like acetylcholinesterase and fumarase are “perfect enzymes,” each enzyme having evolved to the point where nearly every collision with its substrate converts the substrate to a product.
This situation changes drastically when the polysaccharide binds to lysozyme. The active site of lysozyme, because its substrate is a polymer, is a long groove that holds six linked sugars at the same time. As soon as the polysaccharide binds to form an enzyme–substrate complex, the enzyme cuts the polysaccharide by adding a water molecule across one of its sugar–sugar bonds. The product chains are then quickly released, freeing the enzyme for further cycles of reaction (Figure 3–48).
Figure 3–48The overall reaction catalyzed by lysozyme. (A) The enzyme lysozyme (E) catalyzes the cutting of a polysaccharide chain, which is its substrate (S). The enzyme first binds to the chain to form an enzyme–substrate complex (ES) and then catalyzes the cleavage of a specific covalent bond in the backbone of the polysaccharide, forming an enzyme–product complex (EP) that rapidly dissociates. Release of the severed chain (the products P) leaves the enzyme free to act on another substrate molecule. (B) A space-filling model of the lysozyme molecule bound to a short length of polysaccharide chain before cleavage (Movie 3.8). (PDB code: 3AB6.)
An impressive increase in hydrolysis rate is possible because conditions are created in the microenvironment of the lysozyme active site that greatly reduce the activation energy necessary for the hydrolysis to take place. In particular, lysozyme distorts one of the two sugars connected by the bond to be broken from its normal, most stable conformation. The bond to be broken is also held close to two amino acids with acidic side chains (a glutamic acid and an aspartic acid) that participate directly in the reaction. Figure 3–49 highlights the three central steps in this enzymatically catalyzed reaction, which occurs millions of times faster than uncatalyzed hydrolysis.
Figure 3–49Events at the active site of lysozyme. The top left and top right drawings show the free substrate and the free products, respectively. The other three drawings show the sequential events at the enzyme active site, where a sugar–sugar covalent bond is bent and then broken. Note the change in the conformation of sugar D in the enzyme–substrate complex compared with its conformation in the free substrate. This changed conformation favors the formation of the transition state shown in the middle panel, greatly lowering the activation energy that is required for the reaction. This reaction, and the structure of lysozyme bound to its product, are shown in Movie 3.8 and Movie 3.9. (Based on D.J. Vocadlo et al., Nature 412:835–838, 2001.)
Other enzymes use similar mechanisms to lower activation energies and speed up the reactions they catalyze. In reactions involving two or more reactants, the active site also acts like a template, or mold, that brings the substrates together in the proper orientation for a reaction to occur between them (Figure 3–50A). As we saw for lysozyme, the active site of an enzyme contains precisely positioned atoms that speed up a reaction by using charged groups to alter the distribution of electrons in the substrates (Figure 3–50B). And as we have also seen, when a substrate binds to an enzyme, bonds in the substrate are often distorted, changing the substrate shape. These changes drive a substrate toward a particular transition state (Figure 3–50C). Finally, like lysozyme, many enzymes participate intimately in the reaction by transiently forming a covalent bond between the substrate and a side chain of the enzyme. Subsequent steps in the reaction restore the side chain to its original state, so that the enzyme remains unchanged after the reaction (see also Figure 2–47).
Figure 3–50Some general strategies used for enzyme catalysis. (A) Holding substrates together in a precise alignment. (B) Charge stabilization of reaction intermediates. (C) Applying forces that distort bonds in the substrate to increase the rate of a particular reaction.
Tightly Bound Small Molecules Add Extra Functions to Proteins
Although we have emphasized the versatility of enzymes—and proteins in general—as chains of amino acids that perform remarkable functions, there are many instances in which the amino acids by themselves are not enough. Just as humans employ tools to enhance and extend the capabilities of their hands, enzymes and other proteins often use small nonprotein molecules to perform functions that would be difficult or impossible to do with amino acids alone. Thus, enzymes frequently have a small molecule or metal atom tightly associated with their active site that assists with their catalytic function. Carboxypeptidase, for example, an enzyme that cuts polypeptide chains, carries a tightly bound zinc ion in its active site. During the cleavage of a peptide bond by carboxypeptidase, the zinc ion forms a transient bond with one of the substrate atoms, thereby assisting the hydrolysis reaction. In other enzymes, a small organic molecule serves a similar purpose. Such organic molecules are often referred to as coenzymes. An example is biotin, which is found in enzymes that transfer a carboxylate group (–COO–) from one molecule to another (see Figure 2–40). Biotin participates in these reactions by forming a transient covalent bond to the –COO– group to be transferred, being better suited to this function than any of the amino acids used to make proteins. Because it cannot be synthesized by humans, and must therefore be supplied in small quantities in our diet, biotin is a vitamin. Many other coenzymes are either vitamins or derivatives of vitamins (Table 3–2).
TABLE 3–2 Many Vitamin Derivatives Are Critical Coenzymes for Human Cells
Vitamin
Coenzyme
Enzyme-catalyzed reactions requiring these coenzymes
Thiamine (vitamin B1)
Thiamine pyrophosphate
Activation and transfer of aldehydes
Riboflavin (vitamin B2)
FADH
Oxidation–reduction
Niacin
NADH, NADPH
Oxidation–reduction
Pantothenic acid
Coenzyme A
Acyl group activation and transfer
Pyridoxine
Pyridoxal phosphate
Amino acid activation; also glycogen phosphorylase
Biotin
Biotin
CO2 activation and transfer
Lipoic acid
Lipoamide
Acyl group activation; oxidation–reduction
Folic acid
Tetrahydrofolate
Activation and transfer of single carbon groups
Vitamin B12
Cobalamin coenzymes
Isomerization and methyl group transfers
Other proteins also frequently require specific small-molecule adjuncts to function properly. Thus, the signal receptor protein rhodopsin, which is made by the photoreceptor cells in the retina, detects light by means of a small molecule, retinal, embedded in the protein (Figure 3–51A). Retinal, which is derived from vitamin A, changes its shape when it absorbs a photon of light, and this change causes the protein to trigger a cascade of enzymatic reactions that eventually lead to an electrical signal being carried to the brain.
Another example of a protein with a nonprotein portion is hemoglobin (see Figure 3–20). Each molecule of hemoglobin carries four heme groups, ring-shaped molecules each with a single central iron atom (Figure 3–51B). Heme gives hemoglobin (and blood) its red color. By binding reversibly to oxygen gas through its iron atom, heme enables hemoglobin to pick up oxygen in the lungs and release it in the tissues.
Figure 3–51Retinal and heme. (A) The structure of retinal, the light-sensitive molecule attached to rhodopsin in the eye, showing its isomerization when it absorbs light. (B) The structure of a heme group. The carbon-containing heme ring is red and the iron atom at its center is orange. A heme group is tightly bound to each of the four polypeptide chains in hemoglobin, the oxygen-carrying protein whose structure is shown in Figure 3–20.
Sometimes these small molecules are attached covalently and permanently to their protein, thereby becoming an integral part of the protein molecule itself. We shall see in Chapter 10 that proteins are often anchored to cell membranes through covalently attached lipid molecules. And membrane proteins exposed on the surface of the cell, as well as proteins secreted outside the cell, are often modified by the covalent addition of sugars and oligosaccharides.
The Cell Regulates the Catalytic Activities of Its Enzymes
A living cell contains thousands of enzymes, many of which operate at the same time and in the same small volume of the cytosol. By their catalytic action, these enzymes generate a complex web of metabolic pathways, each composed of chains of chemical reactions in which the product of one enzyme becomes the substrate of the next. In this maze of pathways, there are many branch points (nodes) where different enzymes compete for the same substrate. The system is complex (see Figure 2–62), and elaborate controls are required to regulate when and how rapidly each reaction occurs.
Regulation occurs at many levels. At one level, the cell controls how many molecules of each enzyme it makes by regulating the expression of the gene that encodes that enzyme (discussed in Chapter 7). The cell also controls enzymatic activities by confining sets of enzymes to particular subcellular compartments (discussed in Chapters 12 and 14) or by concentrating them on protein scaffolds (see pp. 170–173). As will be explained later in this chapter, enzymes are also covalently modified to control their activity. The rate of protein destruction by targeted proteolysis represents yet another important regulatory mechanism (see Figure 6–89). But the most general process that adjusts reaction rates operates through a direct, reversible change in the activity of an enzyme in response to the specific small molecules that it binds.
Figure 3–52Feedback inhibition of a single biosynthetic pathway. The end product Z inhibits the first enzyme that is unique to its synthesis and thereby controls its own level in the cell. This is an example of negative regulation.
The most common type of control occurs when an enzyme binds a molecule that is not a substrate to a special regulatory site outside the active site, thereby altering the rate at which the enzyme converts its substrates to products. For example, in feedback inhibition, a product produced late in a reaction pathway inhibits an enzyme that acts earlier in the pathway. Thus, whenever large quantities of the final product begin to accumulate, this product binds to the enzyme and slows down its catalytic action, thereby limiting the further entry of substrates into that reaction pathway (Figure 3–52). Where pathways branch or intersect, there are usually multiple points of control by different final products, each of which works to regulate its own synthesis (Figure 3–53). Feedback inhibition can work almost instantaneously, and it is rapidly reversed when the level of the product falls.
Figure 3–53Multiple feedback inhibition. In this example, which shows the biosynthetic pathways for four different amino acids in bacteria, the red lines indicate positions at which products feed back to inhibit enzymes. Each amino acid controls the first enzyme specific to its own synthesis, thereby controlling its own levels and avoiding a wasteful or even dangerous buildup of intermediates. The products can also separately inhibit the initial set of reactions common to all the syntheses; in this case, three different enzymes catalyze the initial reaction, each inhibited by a different product.
Feedback inhibition is negative regulation: it prevents an enzyme from acting. Enzymes can also be subject to positive regulation, in which a regulatory molecule stimulates the enzyme’s activity rather than shutting the enzyme down. Positive regulation occurs when a product in one branch of the metabolic network stimulates the activity of an enzyme in another pathway. As one example, the accumulation of ADP activates several enzymes involved in the oxidation of sugar molecules, thereby stimulating the cell to convert more ADP to ATP.
Allosteric Enzymes Have Two or More Binding Sites That Interact
A striking feature of both positive and negative feedback regulation is that the regulatory molecule often has a shape totally different from the shape of the substrate of the enzyme. This is why the effect on a protein is termed allostery (from the Greek words allos, meaning “other,” and stereos, meaning “solid” or “three-dimensional”). As biologists learned more about feedback regulation, they recognized that the enzymes involved must have at least two different binding sites on their surface—an active site that recognizes the substrates, and a regulatory site that recognizes a regulatory molecule. These two sites must somehow communicate, so that the catalytic events at the active site can be influenced by the binding of the regulatory molecule at its separate site on the protein’s surface.
The interaction between separated sites on a protein molecule is now known to depend on a conformational change in the protein: binding at one of the sites causes a shift from one folded shape to a slightly different folded shape. During feedback inhibition, for example, the binding of an inhibitor at one site on the protein causes the protein to shift to a conformation that incapacitates its active site located elsewhere in the protein.
It is thought that most protein molecules are allosteric. They can adopt many slightly different conformations, and a shift from one to another caused by the binding of a ligand can alter their activity. This is true not only for enzymes but also for many other proteins, including receptors, structural proteins, and motor proteins. In all instances of allosteric regulation, each conformation of the protein has somewhat different surface contours, and the protein’s binding sites for ligands are altered when the protein changes shape. Importantly, as we discuss next, each ligand will stabilize the conformation that it binds to most strongly, and thus—at high enough concentrations—will tend to “switch” the protein toward the conformation that has a high affinity for that ligand.
Two Ligands Whose Binding Sites Are Coupled Must Reciprocally Affect Each Other’s Binding
The effects of ligand binding on a protein follow from a fundamental chemical principle known as linkage. Suppose, for example, that a protein that binds glucose also binds another molecule, X, at a distant site on the protein’s surface. If the binding site for X changes shape as part of the conformational change in the protein induced by glucose binding, the binding sites for X and for glucose are said to be coupled. Whenever two ligands prefer to bind to the same conformation of an allosteric protein, it follows from basic thermodynamic principles that each ligand must increase the affinity of the protein for the other. For example, if the shift of a protein to a conformation that binds glucose best also causes the binding site for X to fit X better, then the protein will bind glucose more tightly when X is present than when X is absent. In other words, X will positively regulate the protein’s binding of glucose (Figure 3–54).
Figure 3–54Positive regulation caused by conformational coupling between two separate binding sites. In this example, both glucose and molecule X bind best to the closed conformation of a protein with two domains. Because both glucose and molecule X drive the protein toward its closed conformation, each ligand helps the other to bind. Glucose and molecule X are therefore said to bind cooperatively to the protein.
Conversely, linkage operates in a negative way if two ligands prefer to bind to different conformations of the same protein. In this case, the binding of the first ligand discourages the binding of the second ligand. Thus, if a shape change caused by glucose binding decreases the affinity of a protein for molecule X, the binding of X must also decrease the protein’s affinity for glucose (Figure 3–55). The linkage relationship is quantitatively reciprocal, so that, for example, if glucose has a very large effect on the binding of X, X has a very large effect on the binding of glucose.
Figure 3–55Negative regulation caused by conformational coupling between two separate binding sites. The scheme here resembles that in the previous figure, but here molecule X prefers the open conformation, while glucose prefers the closed conformation. Because glucose and molecule X drive the protein toward opposite conformations (closed and open, respectively), the presence of either ligand interferes with the binding of the other.
The relationships shown in Figures 3–54 and 3–55 apply to all proteins, and they underlie all of cell biology. The principle seems so obvious in retrospect that we now take it for granted. But the discovery of linkage in studies of a few enzymes in the 1950s, followed by an extensive analysis of allosteric mechanisms in proteins in the early 1960s, had a revolutionary effect on our understanding of biology. Because molecule X in these examples binds at a site on the enzyme that is distinct from the site where catalysis occurs, it need not have any chemical relationship to the substrate that binds at the active site. Moreover, as we have just seen, for enzymes that are regulated in this way, molecule X can either turn the enzyme on (positive regulation) or turn it off (negative regulation). By such a mechanism, allosteric proteins serve as general switches that, in principle, can allow one molecule in a cell to affect the fate of any other.
Symmetrical Protein Assemblies Produce Cooperative Allosteric Transitions
A single-subunit enzyme that is regulated by negative feedback can at most decrease from 90% to about 10% activity in response to a 100-fold increase in the concentration of an inhibitory ligand that it binds (Figure 3–56, red line). Responses of this type are apparently not sharp enough for optimal cell regulation, and most enzymes that are turned on or off by ligand binding consist of symmetrical assemblies of identical subunits. With this arrangement, the binding of a molecule of ligand to a single site on one subunit can promote an allosteric change in the entire assembly that helps the neighboring subunits bind the same ligand. As a result, a cooperative allosteric transition occurs (Figure 3–56, blue line), allowing a relatively small change in ligand concentration in the cell to switch the whole assembly from an almost fully active to an almost fully inactive conformation (or vice versa).
Figure 3–56Enzyme activity versus the concentration of inhibitory ligand for single-subunit and multisubunit allosteric enzymes. For an enzyme with a single subunit (red line), a drop from 90% enzyme activity to 10% activity (indicated by the two dots on the curve) requires a 100-fold increase in the concentration of inhibitor. The enzyme activity is calculated from the simple equilibrium relationship K= [IP]/[I][P], where P is active protein, I is inhibitor, and IP is the inactive protein bound to inhibitor. An identical curve applies to any simple binding interaction between two molecules, A and B. In contrast, a multisubunit allosteric enzyme can respond in a switchlike manner to a change in ligand concentration: the steep response is caused by a cooperative binding of the ligand molecules, as explained in Figure 3–57. Here, the green line represents the idealized result expected for the cooperative binding of two inhibitory ligand molecules to an allosteric enzyme with two subunits, and the blue line shows the idealized response of an enzyme with four subunits. As indicated by the two dots on each of these curves, the more complex enzymes drop from 90% to 10% activity over a much narrower range of inhibitor concentration than does the enzyme composed of a single subunit.
The principles involved in a cooperative “all-or-none” transition are the same for all proteins, whether or not they are enzymes. Thus, for example, they are critical for the efficient uptake and release of O2 by hemoglobin in our blood. But they are perhaps easiest to visualize for an enzyme that forms a symmetrical dimer. In the example shown in Figure 3–57, the first molecule of an inhibitory ligand binds with great difficulty because its binding disrupts an energetically favorable interaction between the two identical monomers in the dimer. A second molecule of inhibitory ligand now binds more easily, however, because its binding restores the energetically favorable monomer–monomer contacts of a symmetrical dimer (this also completely inactivates the enzyme).
Figure 3–57A cooperative allosteric transition in an enzyme composed of two identical subunits. This diagram illustrates how the conformation of one subunit can influence that of its neighbor. The binding of a single molecule of an inhibitory ligand (orange) to one subunit of the enzyme occurs with difficulty because it changes the conformation of this subunit and thereby disrupts the energetically favorable interactions in the symmetrical enzyme. Once this conformational change has occurred, however, the free energy gained by restoring the symmetrical pairing interaction between the two subunits makes it especially easy for the second subunit to bind the inhibitory ligand and undergo the same conformational change. Because the binding of the first molecule of ligand increases the affinity with which the other subunit binds the same ligand, the response of the enzyme to changes in the concentration of the ligand is much steeper than the response of an enzyme with only one subunit (see Figure 3–56 and Movie 3.10).
As an alternative to this induced fit model for a cooperative allosteric transition, we can view such a symmetrical enzyme as having only two possible conformations, corresponding to the “enzyme on” and “enzyme off” structures in Figure 3–57. In this view, ligand binding perturbs an all-or-none equilibrium between these two states, thereby changing the proportion of active molecules. Both models represent true and useful concepts.
Many Changes in Proteins Are Driven by Protein Phosphorylation
Proteins are regulated by more than the reversible binding of other molecules. A second method that eukaryotic cells use extensively to regulate a protein’s function is the covalent addition of a smaller molecule to one or more of its amino acid side chains. The most common such regulatory modification in higher eukaryotes is the addition of a phosphate group. We shall therefore use protein phosphorylation to illustrate some of the general principles involved in the control of protein function through the covalent modification of amino acid side chains.
A phosphorylation event (by a kinase) can affect the protein that is modified in three important ways. First, because each phosphate group carries two negative charges, the enzyme-catalyzed addition of a phosphate group to a protein can cause a major conformational change in the protein by, for example, attracting a cluster of positively charged amino acid side chains. This can, in turn, affect the binding of ligands elsewhere on the protein surface, dramatically changing the protein’s activity. When a second enzyme (called a phosphatase) removes the phosphate group, the protein returns to its original conformation and restores its initial activity.
Second, an attached phosphate group can form part of a structure that the binding sites of other proteins recognize. As previously discussed, the SH2 domain binds to a short peptide sequence containing a phosphorylated tyrosine side chain (see Figure 3–38B). More than 10 other common domains provide binding sites for attaching their protein to phosphorylated peptides in other protein molecules, each recognizing a phosphorylated amino acid side chain in a different protein context. Third, the addition of a phosphate group can mask a binding site that otherwise holds two proteins together, and thereby disrupt protein–protein interactions. As a result of the last two effects, protein phosphorylation and dephosphorylation very often drive the regulated assembly and disassembly of protein complexes.
Reversible protein phosphorylation controls the activity, structure, and cellular localization of enzymes and many other types of proteins in eukaryotic cells. In fact, this regulation is so extensive that more than one-third of the 10,000 or so proteins in a typical mammalian cell are thought to be phosphorylated at any given time—many with more than one phosphate.
As might be expected, the addition and removal of phosphate groups from specific proteins often occur in response to signals that specify some change in a cell’s state. For example, the complicated series of events that takes place as a eukaryotic cell divides is largely timed in this way (discussed in Chapter 17), and many of the signals mediating cell–cell interactions are relayed from the plasma membrane to the nucleus by a cascade of protein phosphorylation events (discussed in Chapter 15).
A Eukaryotic Cell Contains a Large Collection of Protein Kinases and Protein Phosphatases
Protein phosphorylation involves the enzyme-catalyzed transfer of the terminal phosphate group of an ATP molecule to the hydroxyl group on a serine, threonine, or tyrosine side chain of the protein (Figure 3–58). A protein kinase catalyzes this reaction, and the reaction is essentially unidirectional because of the large amount of free energy released when the phosphate–phosphate bond in ATP is broken to produce ADP (discussed in Chapter 2). A protein phosphatase catalyzes the reverse reaction of phosphate removal, or dephosphorylation. Cells contain hundreds of different protein kinases, each responsible for phosphorylating a different protein or set of proteins. There are also many different protein phosphatases; some are highly specific and remove phosphate groups from only one or a few proteins, whereas others act on a broad range of proteins and are targeted to specific substrates by regulatory subunits. The state of phosphorylation of a protein at any moment, and thus its activity, depends on the relative activities of the protein kinases and phosphatases that modify it.
Figure 3–58Protein phosphorylation. Many thousands of proteins in a typical eukaryotic cell are modified by the covalent addition of a phosphate group. (A) The general reaction transfers a phosphate group from ATP to an amino acid side chain of the target protein, catalyzed by a protein kinase. Removal of the phosphate group is catalyzed by a second enzyme, a protein phosphatase. In this example, the phosphate is added to a serine side chain; in other cases, the phosphate is instead linked to the –OH group of a threonine or a tyrosine in the protein. (B) The phosphorylation of a protein by a protein kinase can either increase or decrease the protein’s activity, depending on the site of phosphorylation and the structure of the protein.
The protein kinases that phosphorylate proteins in eukaryotic cells belong to a very large family of enzymes that share a catalytic (kinase) sequence of about 290 amino acids. The various family members contain different amino acid sequences on either end of the kinase sequence (for example, see Figure 3–11) and often have short amino acid sequences inserted into loops within it. Some of these additional amino acid sequences enable each kinase to recognize the specific set of proteins it phosphorylates or to bind to structures that localize it in specific regions of the cell. Other parts of the protein regulate the activity of each kinase, so it can be turned on and off in response to different specific signals, as described below.
By comparing the number of amino acid sequence differences between the various members of a protein family, we can construct an “evolutionary tree” that is thought to reflect the pattern of gene duplication and divergence that gave rise to the family. Figure 3–59 shows an evolutionary tree for protein kinases. Kinases with related functions are often located on nearby branches of the tree: the protein kinases involved in cell signaling that phosphorylate tyrosine side chains, for example, are all clustered in the top left corner of the tree. The other kinases shown phosphorylate either a serine or a threonine side chain, and many are organized into clusters that seem to reflect their function—in transmembrane signal transduction, intracellular signal amplification, cell-cycle control, and so on.
Figure 3–59An evolutionary tree of selected protein kinases. A higher eukaryotic cell contains hundreds of such enzymes, and the human genome codes for more than 500. Note that only some of these, those discussed in this book, are shown.
As a result of the combined activities of protein kinases and protein phosphatases, the phosphate groups on proteins are continually turning over—being added and then rapidly removed. Such phosphorylation cycles may seem wasteful, but they are important in allowing the phosphorylated proteins to switch rapidly from one state to another. In fact, the more rapid this cycle is “turning,” the faster a population of protein molecules can change its state of phosphorylation in response to a sudden change in its phosphorylation rate (see Figure 15–15). The energy required to drive this phosphorylation cycle is derived from the free energy of ATP hydrolysis, one molecule of which is consumed for each phosphorylation event.
The Regulation of the Src Protein Kinase Reveals How a Protein Can Function as a Microprocessor
The hundreds of different protein kinases in a eukaryotic cell are organized into complex networks of signaling pathways that help to coordinate the cell’s activities, drive the cell cycle, and relay signals into the cell from the cell’s environment. Many of the extracellular signals involved need to be both integrated and amplified by the cell. Individual protein kinases (and other signaling proteins) serve as input–output devices, or “microprocessors,” in the integration process. An important part of the input to these signal-processing proteins comes from the control that is exerted by phosphates added and removed from them by protein kinases and protein phosphatases, respectively.
The Src family of protein kinases (see Figure 3–11) exhibits such behavior. The Src protein (pronounced “sarc” and named for the type of tumor, a sarcoma, that its deregulation can cause) was the first tyrosine kinase to be discovered. It is now known to be part of a subfamily of nine very similar protein kinases, which are found only in multicellular animals. As indicated by the evolutionary tree in Figure 3–59, sequence comparisons suggest that tyrosine kinases as a group were a relatively late innovation that branched off from the serine/threonine kinases, with the Src subfamily being only one subgroup of the tyrosine kinases created in this way.
The Src protein and its relatives contain a short N-terminal region that becomes covalently linked to a strongly hydrophobic fatty acid, which anchors the kinase at the cytoplasmic face of the plasma membrane. Next along the linear sequence of amino acids come two peptide-binding domains, a Src homology 3 (SH3) domain and an SH2 domain, followed by the kinase catalytic domain (Figure 3–60). These kinases normally exist in an inactive conformation, in which a phosphorylated tyrosine near the C-terminus is bound to the SH2 domain, and the SH3 domain is bound to an internal peptide in a way that distorts the active site of the enzyme and helps to render it inactive.
Figure 3–60The domain structure of the Src family of protein kinases, mapped along the amino acid sequence. For the three-dimensional structure of Src, see Figure 3–11.
As shown in Figure 3–61, turning the kinase on involves at least two specific inputs: removal of the C-terminal phosphate and the binding of the SH3 domain by a specific activating protein. In this way, the activation of the Src kinase signals the completion of a particular set of separate upstream events (Figure 3–62). Thus, the Src family of protein kinases serves as specific signal integrators, contributing to the web of information-processing events that enable the cell to compute useful responses to a complex set of different conditions.
Figure 3–61The activation of a Src-type protein kinase by two sequential events. As described in the text, the requirement for multiple upstream events to trigger these processes allows the kinase to serve as a signal integrator (Movie 3.11). (Adapted from S.C. Harrison, Cell 112:737–740, 2003.)Figure 3–62How a Src-type protein kinase acts as a signal-integrating device. A disruption of the inhibitory interaction illustrated for the SH3 domain (green) occurs when its binding to the indicated orange linker region is replaced with its higher-affinity binding to an activating ligand.
Regulatory GTP-binding Proteins Are Switched On and Off by the Gain and Loss of a Phosphate Group
Eukaryotic cells have a second way to regulate protein activity by phosphate addition and removal. In this case, however, the phosphate is not enzymatically transferred from ATP to the protein. Instead, the phosphate is part of a guanine nucleotide—guanosine triphosphate (GTP)—that binds tightly to various types of GTP-binding proteins. These proteins, also called GTPases, bind to other proteins to regulate their activities. They serve as molecular switches: GTP-binding proteins are in their “on” conformation when GTP is bound, but they can hydrolyze this GTP to GDP—which releases a phosphate and flips the protein to its “off” conformation. As with protein phosphorylation, this process is reversible: the active conformation is regained by dissociation of the GDP, followed by the rapid binding of a fresh molecule of GTP (Figure 3–63).
Figure 3–63Many different GTP-binding proteins function as molecular switches. The activity of a GTP-binding protein (also called a GTPase) generally requires the presence of a tightly bound GTP molecule (switch “on”). Hydrolysis of this GTP molecule by the GTP-binding protein—at a rate that can be regulated—produces GDP and inorganic phosphate, and it causes the protein to convert to a different, usually inactive, conformation (switch “off”). Resetting the switch to “on” requires that the tightly bound GDP dissociate. This is a slow step, and the dissociation of GDP, which is followed by its rapid replacement by GTP, is controlled by cell signals (see Figure 15–8).
Hundreds of different GTP-binding proteins function as such molecular switches in cells. They all contain variations of the same globular domain that undergoes a conformational change when its tightly bound GTP is hydrolyzed to GDP. The three-dimensional structure of a prototypical member of this family, the monomeric GTPase called Ras that plays important roles in cell signaling, is shown in Figure 3–64.
Figure 3–64The structure of the Ras protein in its GTP-bound form. This monomeric GTPase illustrates the structure of a GTP-binding domain, which is present in a large family of GTP-binding proteins. The red regions change their conformation when the GTP molecule is hydrolyzed to GDP and inorganic phosphate by the protein; the GDP remains bound to the protein, while the inorganic phosphate is released. The special role of the switch helix in proteins related to Ras is explained in the text (see Figure 3–68 and Movie 15.7). (PDB code: 121P.)
The crucial role that GTP-binding proteins play in intracellular signaling pathways is discussed in detail in Chapter 15.
Proteins Can Be Regulated by the Covalent Addition of Other Proteins
Cells contain a special family of small proteins whose members are covalently attached to many other proteins to determine the activity or fate of the second protein. In each case, the carboxyl end of the small protein becomes linked to the amino group of a lysine side chain of a target protein through an isopeptide bond. The first such protein discovered, and the most abundantly used, is ubiquitin (Figure 3–65A). Ubiquitin can be covalently attached to target proteins in a variety of ways, each of which has a different meaning for cells. The major form of ubiquitin addition produces polyubiquitin chains in which—once the first ubiquitin molecule is attached to the target—each subsequent ubiquitin molecule links to Lys48 of the previous ubiquitin, creating a chain of Lys48-linked ubiquitins that are attached to a single lysine side chain of the target protein. This form of polyubiquitin directs the target protein to the interior of a proteasome, where it is digested to small peptides (see Figure 6–87). In other circumstances, only single molecules of ubiquitin are added to proteins. In addition, some target proteins are modified with a different type of polyubiquitin chain. These modifications have different functional consequences for the protein that is targeted (Figure 3–65B).
Figure 3–65The marking of proteins by ubiquitin. (A) The three-dimensional structure of ubiquitin, a small protein of 76 amino acids. A family of special enzymes couples its carboxyl end to the amino group of a lysine side chain in a target protein molecule, forming an isopeptide bond. (B) Some modification patterns that have specific meanings to the cell. Note that the two types of polyubiquitylation differ in the way the ubiquitin molecules are linked together. Linkage through Lys48 signifies degradation by the proteasome (see Figure 6–87), whereas that through Lys63 has other meanings. Ubiquitin markings are “read” by proteins that specifically recognize each type of modification.
Related structures are created when a different member of the ubiquitin family, such as SUMO (small ubiquitin-related modifier), is covalently attached to a lysine side chain of target proteins. Not surprisingly, all such modifications are reversible. Cells contain sets of ubiquitylating and deubiquitylating (and sumoylating and desumoylating) enzymes that manipulate these covalent adducts, thereby playing roles analogous to the protein kinases and protein phosphatases that add and remove phosphates from protein side chains.
An Elaborate Ubiquitin-conjugating System Is Used to Mark Proteins
How do cells select target proteins for ubiquitin addition? As an initial step, the carboxyl end of ubiquitin needs to be activated. This activation is accomplished when a protein called a ubiquitin-activating enzyme (E1) uses ATP hydrolysis energy to attach ubiquitin to itself through a high-energy covalent bond (a thioester). E1 then passes this activated ubiquitin to one of a set of ubiquitin-conjugating (E2) enzymes, each of which acts in conjunction with a set of accessory (E3) proteins called ubiquitin ligases that select the target proteins to be modified. There are roughly 30 structurally similar but distinct E2 enzymes in mammals and hundreds of different E3 proteins that form complexes with specific E2 enzymes.
Figure 3–66 illustrates the process used to mark proteins for proteasomal degradation. [Similar mechanisms are used to attach ubiquitin (and SUMO) to other types of target proteins.] Here, the ubiquitin ligase binds to specific degradation signals, called degrons, in protein substrates, thereby helping E2 to form a polyubiquitin chain linked to a lysine of the substrate protein. This polyubiquitin chain on a target protein will then be recognized by a specific receptor in the proteasome, causing the target protein to be rapidly destroyed. Distinct ubiquitin ligases recognize different degradation signals, thereby targeting distinct subsets of intracellular proteins for destruction, often in response to specific signals (see Figure 6–89).
Figure 3–66How ubiquitin is added to proteins. (A) Ubiquitin activations. The C-terminus of ubiquitin is initially activated by being linked via a high-energy thioester bond to a cysteine side chain on the E1 protein. This reaction requires ATP, and it proceeds via a covalent AMP–ubiquitin intermediate. The activated ubiquitin on E1, also known as the ubiquitin-activating enzyme, is then transferred to the cysteine on an E2 molecule. (B) The addition of a polyubiquitin chain to a target protein. In a mammalian cell, there are several hundred distinct E2–E3 complexes. The E2s are called ubiquitin-conjugating enzymes. The E3s are referred to as ubiquitin ligases. (Adapted from D.R. Knighton et al., Science 253:407–414, 1991.)
Protein Complexes with Interchangeable Parts Make Efficient Use of Genetic Information
Controlled protein degradation is critical for cells, and we will describe the structure and function of one of the families of E3 proteins that adds polyubiquitin chains to target proteins in order to illustrate a general principle: how the cell makes use of interchangeable parts to diversify its many protein complexes.
The SCF ubiquitin ligase is a C-shaped structure that is formed from five protein subunits, the largest of which serves as a scaffold on which the rest of the complex is built. The structure underlies a remarkable mechanism (Figure 3–67). At one end of the C is an E2 ubiquitin–conjugating enzyme. At the other end is a substrate-binding arm, a subunit known as an F-box protein. These two subunits are separated by a gap of about 5 nm. When this protein complex is activated, the F-box protein binds to a specific site on a target protein, positioning the protein in the gap so that some of its lysine side chains contact the ubiquitin-conjugating enzyme. The enzyme can then catalyze repeated additions of ubiquitin to these lysines (see Figure 3–67C), producing the polyubiquitin chains that mark its target proteins for destruction in a proteasome.
Figure 3–67The structure and mode of action of a ubiquitin ligase. (A) The structure of the five-protein SCF ubiquitin ligase complex that includes an E2 ubiquitin-conjugating enzyme. Four proteins form the E3 portion. The protein denoted here as adaptor protein 1 is the Rbx1/Hrt1 protein, adaptor protein 2 is the Skp1 protein, and cullin is the Cul1 protein. One of the many different F-box proteins completes the complex. (B) Comparison of the same complex with two different substrate-binding arms, the F-box proteins Skp2 (top) and β-trCP1 (bottom), respectively. (C) The binding and ubiquitylation of a target protein by the SCF ubiquitin ligase. If, as indicated, a chain of ubiquitin molecules is added to the same lysine of the target protein, that protein is marked for rapid destruction by the proteasome. (D) Comparison of SCF (bottom) with a low-resolution electron microscopy structure of a ubiquitin ligase called the anaphase-promoting complex (APC/C; top) at the same scale. The APC/C is a large, 15-protein complex. As discussed in Chapter 17, its ubiquitylations control the late stages of mitosis. It is distantly related to SCF and contains a cullin subunit (green) that lies along the side of the complex at right, only partly visible in this view. E2 proteins are not shown here, but their binding sites are indicated in orange, along with substrate-binding sites in purple. (A and B, adapted from G. Wu et al., Mol. Cell 11:1445–1456, 2003. D, adapted from P. da Fonseca et al., Nature 470:274–278, 2011.)
In this manner, specific proteins are targeted for rapid destruction in response to specific signals, thereby helping to drive the cell cycle (discussed in Chapter 17). The timing of the destruction often involves creating a specific pattern of phosphorylation on the target protein that is required for its recognition by the F-box subunit. It also requires the activation of an SCF-like ubiquitin ligase that carries the appropriate substrate-binding arm. Many of these arms (the F-box subunits) are interchangeable in the protein complex (see Figure 3–67B), and there are more than 70 human genes that encode them.
As emphasized previously, once a successful protein has evolved, its genetic information tends to be duplicated to produce a family of related proteins. Thus, for example, not only are there many F-box proteins—making possible the recognition of different sets of target proteins—but there is also a family of scaffolds (known as cullins) that give rise to a family of SCF-like ubiquitin ligases.
A protein machine like the SCF ubiquitin ligase, with its interchangeable parts, makes economical use of the genetic information in cells. It also creates opportunities for “rapid” evolution, inasmuch as new functions can evolve for the entire complex simply by producing an alternative version of one of its subunits.
Ubiquitin ligases form a diverse family of protein complexes. Some of these complexes are far larger and more complicated than SCF, but their underlying enzymatic function remains the same (see Figure 3–67D).
A GTP-binding Protein Shows How Large Protein Movements Can Be Generated from Small Ones
Detailed structures obtained for one of the GTP-binding protein family members, the EF-Tu protein, provide a good example of how allosteric changes in protein conformations can produce large movements by amplifying a small, local conformational change. As will be discussed in Chapter 6, EF-Tu is an abundant molecule that serves as an elongation factor (hence the EF) in protein synthesis, loading each aminoacyl-tRNA molecule onto the ribosome. EF-Tu contains a Ras-like domain (see Figure 3–64), and the tRNA molecule forms a tight complex with its GTP-bound form. For the tRNA molecule to transfer its amino acid to the growing polypeptide chain requires that the GTP bound to EF-Tu be hydrolyzed, dissociating the EF-Tu from the tRNA. Because this GTP hydrolysis is triggered by a proper fit of the tRNA to the mRNA molecule on the ribosome, the EF-Tu serves to discriminate between correct and incorrect mRNA–tRNA pairings (see Figure 6–69).
By comparing the three-dimensional structure of EF-Tu in its GTP-bound and GDP-bound forms, we can see how the repositioning of the tRNA occurs. The dissociation of the inorganic phosphate group, which follows the reaction GTP → GDP + phosphate, causes a shift of a few tenths of a nanometer at the GTP-binding site, just as it does in the Ras protein. This tiny movement, equivalent to a few times the diameter of a hydrogen atom, causes a conformational change to propagate along a crucial piece of α helix, called the switch helix, in the Ras-like domain of the protein. The switch helix seems to serve as a latch that adheres to a specific site in another domain of the molecule, holding the protein in a “shut” conformation. The conformational change triggered by GTP hydrolysis causes the switch helix to detach, allowing separate domains of the protein to swing apart, through a distance of about 4 nm (Figure 3–68). This releases the tRNA, allowing its attached amino acid to be used for protein synthesis (Figure 3–69).
Figure 3–68The large conformational change in EF-Tu caused by GTP hydrolysis. (A and B) The three-dimensional structure of EF-Tu with GTP bound. The domain at the top has a structure similar to the Ras protein, and its red α helix is the switch helix, which moves after GTP hydrolysis. (C) The change in the conformation of the switch helix in domain 1 allows domains 2 and 3 to rotate as a single unit by about 90 degrees toward the viewer, which releases the tRNA that was bound to this structure (see also Figure 3–69). (A, PDB code: 1EFT; B, courtesy of Mathias Sprinzl and Rolf Hilgenfeld.)Figure 3–69An aminoacyl tRNA molecule bound to EF-Tu. Note how the bound protein blocks the use of the tRNA-linked amino acid (dark green) for protein synthesis until GTP hydrolysis triggers the conformational changes shown in Figure 3–68C, dissociating the protein–tRNA complex. EF-Tu is a bacterial protein; however, a very similar protein exists in eukaryotes, where it is called EF-1 (Movie 3.12). (PDB code: 1B23.)
Notice in this example how cells have exploited a simple chemical change that occurs on the surface of a small protein domain to create a movement 50 times larger. Dramatic shape changes of this type also cause the very large movements that occur in motor proteins, as we discuss next.
Motor Proteins Produce Directional Movement in Cells
We have seen that conformational changes in proteins have a central role in enzyme regulation and cell signaling. We now discuss proteins whose major function is to move other molecules. These motor proteins generate the forces responsible for muscle contraction and the crawling and swimming of cells. Motor proteins also power smaller-scale intracellular movements: they help to move chromosomes to opposite ends of the cell during mitosis (discussed in Chapter 17), to move organelles along molecular tracks within the cell (discussed in Chapter 16), and to move enzymes along a DNA strand during the synthesis of a new DNA molecule (discussed in Chapter 5). All these fundamental processes depend on proteins with moving parts that operate as force-generating machines.
How do these machines work? It is a challenge for cells to use shape changes in proteins to generate persistent movements in a single direction. If, for example, a protein is required to walk along a long cytoskeletal filament, it can do this by undergoing a series of conformational changes, such as those shown in Figure 3–70. But with nothing to drive these changes in an orderly sequence, they are perfectly reversible, and the protein can only wander randomly back and forth along the thread. We can look at this situation in another way. Because the directional movement of a protein does work, the laws of thermodynamics (discussed in Chapter 2) demand that such movement use free energy from some other source (otherwise the protein could be used to make a perpetual motion machine). Therefore, without an input of energy, the protein molecule can only wander aimlessly.
Figure 3–70Changes in conformation can cause a protein to “walk” along a cytoskeletal filament, driven by its constant collisions with other molecules (thermal energy). This protein cycles between three different conformations (A, B, and C) as it moves along the filament. But, without an input of energy to drive its movement in a single direction, the protein can only wander randomly back and forth, ultimately getting nowhere.
How can the cell make such a series of conformational changes unidirectional? To force the entire cycle to proceed in one direction, it is enough to make any one of the changes in shape irreversible. Most proteins that are able to walk in one direction for long distances achieve this motion by coupling one of the conformational changes to the hydrolysis of an ATP molecule that is tightly bound to the protein. The mechanism is similar to the one discussed earlier that drives allosteric protein shape changes by GTP hydrolysis. Because ATP (or GTP) hydrolysis releases a great deal of free energy, it is very unlikely that the nucleotide-binding protein will undergo the reverse shape change needed for moving backward—as this would require that it also reverse the ATP hydrolysis by adding a phosphate molecule to ADP to form ATP.
In the model shown in Figure 3–71A, ATP binding shifts a motor protein from conformation 1 to conformation 2. The bound ATP is then hydrolyzed to produce ADP and inorganic phosphate, causing a change from conformation 2 to conformation 3. Finally, the release of the bound ADP and phosphate drives the protein back to conformation 1. Because the energy provided by ATP hydrolysis drives the transition 2 → 3, this series of conformational changes is effectively irreversible. Thus, the entire cycle goes in only one direction, causing the protein molecule to walk continuously to the right in this example.
Figure 3–71How a protein can walk in one direction. (A) An allosteric motor protein driven by ATP hydrolysis. The transition between three different conformations includes a step driven by the hydrolysis of a tightly bound ATP molecule, creating a “unidirectional ratchet” that makes the entire cycle essentially irreversible. By repeated cycles, the protein therefore moves continuously to the right along the thread. (B) Direct visualization of a walking myosin motor protein by high-speed atomic force microscopy; the elapsed time between steps was less than 0.5 sec (see Movie 16.3). (B, adapted from N. Kodera et al., Nature 468:72–76, published 2010 by Macmillan Publishers Ltd. Reproduced with permission of SNCSC.)
Many motor proteins generate directional movement through the use of a similar unidirectional ratchet, including the muscle motor protein myosin, which walks along actin filaments (Figure 3–71B), and the kinesin proteins that walk along microtubules (both discussed in Chapter 16). These movements can be rapid: some of the motor proteins involved in DNA replication (the DNA helicases) propel themselves along a DNA strand at rates as high as 1000 nucleotides per second.
Proteins Often Form Large Complexes That Function as Protein Machines
Large proteins formed from many domains are able to perform more elaborate functions than small, single-domain proteins. But large protein assemblies formed from many protein molecules linked together by noncovalent bonds perform the most impressive tasks. Now that it is possible to reconstruct most biological processes in cell-free systems in the laboratory, it is clear that each of the central processes in a cell—such as DNA replication, protein synthesis, vesicle budding, or transmembrane signaling—is catalyzed by a highly coordinated, linked set of 10 or more proteins. In most such protein machines, energetically favorable reactions such as the hydrolysis of bound nucleoside triphosphates (ATP or GTP) drive an ordered series of conformational changes in one or more of the individual protein subunits, enabling the ensemble of proteins to move in a coordinated way. As a result, each enzyme can be moved directly into position as the machine catalyzes successive reactions in a series (Figure 3–72). This is what occurs, for example, in protein synthesis on a ribosome (an RNA–protein, or macromolecular machine, discussed in Chapter 6)—or in DNA replication, where a large multiprotein complex moves rapidly along the DNA (discussed in Chapter 5).
Figure 3–72Schematic example showing how protein machines can carry out complex functions. These machines are made of individual proteins that collaborate to perform a specific task (Movie 3.13). As in this example, the movement of these proteins is often coordinated by the hydrolysis of a bound nucleotide such as ATP or GTP. Directional allosteric conformational changes of proteins driven in this way often occur in a large protein assembly, thereby allowing directed movements within the complex to coordinate the activities of its individual molecules. (See also Movie 5.5.)
Cells have evolved protein machines for the same reason that humans have invented mechanical and electronic machines. For accomplishing almost any task, manipulations that are spatially and temporally coordinated through linked processes are much more efficient than the use of many separate tools.
The Disordered Regions in Proteins Are Critical for a Set of Different Functions
Scientists have discovered that proteins contain a surprisingly large amount of intrinsically disordered polypeptide chain. Thus, as previously mentioned, it is estimated that about a third of all eukaryotic proteins contain unstructured regions greater than 30 amino acids in length. Some of these regions are formed from only a limited subset of the 20 amino acids and are therefore designated as low-complexity domains. Because many unstructured regions have been conserved in a particular protein over long periods of evolutionary time, their presence must benefit the organisms that contain them. What do these disordered regions do?
Intrinsically disordered regions of proteins often form specific binding sites for other proteins that are of high specificity, as illustrated in Figure 3–73A. In addition, this type of binding interaction is easily controlled. Most protein phosphorylation sites are in intrinsically disordered regions, not in globular domains, and these regions are central to regulatory mechanisms. As one example, the eukaryotic RNA polymerase enzyme that produces mRNAs contains an unstructured C-terminal tail of 200 amino acids that is covalently modified as the RNA polymerase proceeds, thereby attracting specific other proteins to the transcription complex at different times (see Figure 6–23). Disordered regions tend to evolve rapidly, and the type of binding diagrammed in Figure 3–73B facilitates the fine-tuning and evolution of cell signaling networks (see Chapter 15).
Figure 3–73Intrinsically disordered protein sequences provide versatile binding sites. (A) Unstructured regions of polypeptide chain often form binding sites for other proteins. Although these binding events are of high specificity, they are often of low affinity because of the free-energy cost of folding the normally unfolded partner (and they are thus readily reversible). (B) Unstructured regions can be easily modified covalently to change their binding preferences, and they are therefore frequently involved in cell signaling processes. In this schematic, multiple sites of protein phosphorylation are indicated.
A very different type of function is exemplified by elastin, an abundant protein in the extracellular matrix that is formed as a highly disordered polypeptide. Elastin’s relatively loose and unstructured polypeptide chains are covalently cross-linked to produce an elastic meshwork that can be stretched like a rubber band, as illustrated in Figure 3–74. The elastic fibers that result enable skin and other tissues, such as arteries and lungs, to stretch and recoil without tearing.
Figure 3–74Intrinsically disordered protein chains are used to produce elastic structures. The polypeptide chains of the protein elastin are cross-linked together in the extracellular space to form rubberlike, elastic fibers. Each elastin molecule uncoils into a more extended conformation when the fiber is stretched, and it recoils spontaneously as soon as the stretching force is relaxed.
Perhaps most uniquely, intrinsically disordered regions are widely used as tethers to concentrate reactants and thereby accelerate the reactions needed by a cell. For example, within large multienzyme complexes, unstructured regions of polypeptide chain can allow substrates to be carried sequentially between different active sites (Figure 3–75).
Figure 3–75How unstructured regions of polypeptide chain can serve as tethers to allow reaction intermediates to be passed from one active site to another in a large multienzyme complex, the fatty acid synthase in mammals. (A) The locations of seven protein domains with different activities in this 270-kilodalton protein are shown. The numbers refer to the order in which each enzyme domain must function to complete each two-carbon addition step. After multiple cycles of two-carbon addition, the termination domain releases the final product once the desired length of fatty acid has been synthesized. (B) The structure of the dimeric enzyme, with the location of the five active sites in one monomer indicated. (C) How a flexible tether allows the substrate that remains linked to the acyl carrier domain (red) to be passed from one active site to another in each monomer, sequentially elongating and modifying the bound fatty acid intermediate (yellow). The five steps are repeated until the final length of fatty acid chain has been synthesized. (Only steps 1 through 4 are illustrated here.) (Adapted from T. Maier et al., Q. Rev. Biophys. 43:373–422, 2010.)
In their most general tethering role, unstructured regions allow large scaffold proteins with multiple binding sites to concentrate sets of interacting RNA and/or protein molecules at a particular site in a cell, as we discuss next.
Scaffolds Bring Sets of Interacting Macromolecules Together and Concentrate Them in Selected Regions of a Cell
As scientists have learned more of the details of cell biology, they have recognized an increasing degree of sophistication in cell chemistry. We now know that protein machines play a predominant role and that all of their activities—like those of other proteins—are highly regulated. In addition, it has also become clear that these machines are often localized to specific sites in the cell, being assembled and activated only where and when they are needed. As one example, when extracellular signaling molecules bind to receptor proteins in the plasma membrane, the activated receptors often recruit a set of other proteins to the inside surface of the plasma membrane to form a large protein complex that passes the signal on (illustrated and discussed in Chapter 15).
The mechanisms generally involve scaffold proteins that have binding sites for multiple other proteins and/or RNA molecules. Such scaffolds serve both to link together specific sets of interacting macromolecules and to position them at specific locations inside a cell. At one extreme are rigid scaffolds, such as the cullin in SCF ubiquitin ligase (see Figure 3–67). At the other extreme are large, flexible scaffold proteins that create special regions inside the cell that have a unique biochemistry. Networks of such large scaffolds often underlie regions of specialized plasma membrane. For example, the Discs-large protein (Dlg) of about 900 amino acids is concentrated in special regions beneath the plasma membrane in epithelial cells and at synapses. Dlg contains binding sites for at least seven other proteins interspersed with regions of more flexible polypeptide chain. An ancient protein, conserved in organisms as diverse as sponges, worms, flies, and humans, Dlg derives its name from the mutant phenotype of the organism in which it was first discovered. In a Drosophila embryo with a mutation in the Dlg gene, the imaginal disc cells fail to stop proliferating when they should, and they produce unusually large discs whose epithelial cells can form tumors.
Dlg and a large number of similar scaffold proteins are thought to function like the protein that is schematically illustrated in Figure 3–76. By binding a specific set of interacting proteins and/or RNA molecules, these scaffolds can enhance the rate of critical reactions, while also confining them to the particular region of the cell that contains the scaffold. For similar reasons, cells also make extensive use of scaffold RNA molecules, as discussed in Chapter 7.
Figure 3–76How the proximity created by scaffold proteins can greatly speed reactions in a cell. In this example, long unstructured regions of polypeptide chain in a large scaffold protein connect a series of structured domains that bind a set of reacting proteins or RNA molecules. The unstructured regions serve as flexible tethers that greatly speed reaction rates by causing a rapid, random collision of all of the molecules that are bound to the scaffold. (For specific examples of protein tethering, see Figure 3–75 and Figure 16–14; for scaffold RNA molecules, see Figure 7–82.)
Macromolecules Can Self-assemble to Form Biomolecular Condensates
The macromolecular assemblies and protein machines that we have discussed so far are defined by physical interactions that organize individual proteins and nucleic acids at defined positions relative to each other. Each copy of a macromolecular machine generally is built from the same parts and assembled into the same three-dimensional structure. For example, the bacterial ribosome responsible for synthesizing new proteins is built from 55 proteins and three RNA molecules arranged in an invariant complex (see Figure 6–65). Even in the case of protein complexes containing flexible scaffolds (see Figure 3–75), the macromolecular assembly has a characteristic (albeit flexible) conformation.
In contrast, biomolecular condensates are a different type of cellular structure built from proteins (and often RNA) held together by a large number of weak and constantly changing interactions among them. Each condensate is created by at least one scaffold macromolecule (a protein or RNA molecule) that is capable of making multiple independent interactions with either itself or with other macromolecules, which themselves often make multiple interactions. These types of macromolecules are said to be multivalent. Typically, each of the individual interactions among these multivalent proteins and RNAs is very weak, so it forms and breaks frequently. When any one interaction breaks, other interactions at different sites in that macromolecule prevent it from diffusing away and keep the macromolecule locally concentrated. By the time some of these other interactions break, new ones have already formed elsewhere. In this way, all of the proteins within a condensate continually interact with each other, even though the specific set of interactions changes from one moment to another.
Formation of a condensate serves to segregate and concentrate a subset of the cell’s macromolecules into a separate compartment in the cell. In some cases, these macromolecules perform specialized biochemistry within the condensate—forming a biochemical “factory” that efficiently produces a specific product, as for the ribosomes that are produced by the nucleolus. In other cases, sequestration into a condensate can serve as a temporary storage depot for a set of macromolecules while blocking their activity, as for the stress granules that can form when a cell is perturbed.
The disordered, low-complexity domains of proteins are often found to mediate the fluctuating, weak binding interactions that form a condensate, frequently making a major contribution to their formation. In addition, other types of binding can also drive condensate formation (Figure 3–77A). The dynamic, fluctuating interactions within a condensate cause it to behave like a liquid: all of the participating molecules within it jostle around and rapidly exchange their relative positions; in addition, they often exchange rapidly with their equivalents outside the condensate (Figure 3–77B). Because the condensate remains intact and distinct from the surrounding liquid, the process of condensate formation is commonly termed liquid–liquid phase separation or liquid–liquid demixing.
Figure 3–77The multivalent interactions between scaffold macromolecules that drive the formation of biomolecular condensates. (A) Schematic diagram of a biomolecular condensate that contains both RNA and proteins; illustrated are some types of weak binding interactions frequently involved. Note that the low-complexity domains of scaffold proteins are often critical for forming these condensates, and that several different types of binding interactions are known to cause these unstructured regions to adhere to each other. In addition to the stacking of aromatic side chains (phenylalanine is shown), these include ionic attractions, cation–pi interactions, and the formation of kinked cross-beta structures that resemble amyloids. (B) A fluorescence recovery after photobleaching (FRAP) experiment reveals that the protein molecules inside a condensate are mobile. Here the multiple nucleoli in a mammalian cell have been fluorescently labeled by fusing GFP to the scaffold protein fibrillin, and this fibrillin in one of the nucleoli has been bleached with a flash from a focused laser beam. A rapid recovery of fluorescence demonstrates that the fibrillin in this condensate is continually exchanging with the fibrillin molecules in its surroundings. (B, from R.D. Phair and T. Misteli, Nature 404:604–609, 2000. Reproduced with permission of SNCSC.)
A characteristic feature of biomolecular condensates that reflects their dynamic nature is the readily reversible assembly and disassembly of many of these structures. Thus, for example, the nucleolus disappears during mitosis, and it reforms in early interphase by fusion of the initially separate droplets that form on different chromosomes at the start of each interphase (Figure 3–78). Likewise, the DNA repair, DNA replication, and DNA transcription factories in the nucleus appear only where and when each of these processes occurs (Figure 3–79; see also Figure 6–51C).
Figure 3–78Spherical, liquid-droplet-like nucleoli can be seen to fuse in the light microscope. In these experiments, the nucleoli are present inside a nucleus that has been dissected from Xenopus oocytes and placed under oil on a microscope slide. Here, three nucleoli are seen fusing to form a larger biomolecular condensate. A very similar process occurs after each round of division, when small nucleoli initially form on multiple chromosomes but then coalesce to form a single, large nucleolus (see Figure 6–47). (From C.P. Brangwynne et al., Proc. Natl. Acad. Sci. USA 108:4334–4339, 2011. With permission from National Academy of Sciences.)Figure 3–79The formation of a biomolecular condensate in response to DNA damage. Here, a brief irradiation flash from a UV laser has been used to create a narrow line of DNA damage in the interphase nucleus of a mammalian cell. Because the FUS scaffold protein has been fluorescently labeled with GFP, the formation of the liquid-droplet-like DNA repair factories that this scaffold helps to generate can be followed in a living cell. (Adapted from Movie S1 in A. Patel et al., Cell 162:1066–1077, 2015. With permission from Elsevier.)
Classical Studies of Phase Separation Have Relevance for Biomolecular Condensates
A familiar phase-separation process is that between oil and water, which occurs in some salad dressings. A phase separation occurs whenever forming two phases instead of one minimizes the free energy of a mixture, and it requires overcoming the large unfavorable free-energy change caused by the entropic cost of demixing. Thus, in the oil and water example, there are many more ways of distributing the small oil molecules in between water molecules than there are ways of condensing the oil molecules all together. The completely mixed state is by far the most probable, and the act of demixing therefore involves a large unfavorable (negative) entropy change that produces a large unfavorable (positive) change in the ΔG for phase separation (remembering that ΔG=ΔH–TΔS). But because of an even larger, favorable ΔG derived from preventing the oil molecules from disrupting the hydrogen-bonded network of water molecules, the oil and water separate into distinct phases (see Panel 2–2, pp. 96–97).
For large polymers, which include proteins and nucleic acids, the entropic cost of demixing is considerably less than that for an equivalent mass of small molecules. This is because the monomeric subunits of a polymer are already greatly constrained in their possible arrangements through their covalent attachment to other subunits. As a result, a set of relatively weak attractions between the polymer molecules can often provide a large enough favorable free-energy change to drive phase separation—overcoming the unfavorable free-energy change of demixing.
Chemists have developed phase diagrams to describe what happens when chemically synthesized polymers phase-separate (Figure 3–80). As illustrated, when a threshold concentration of a polymer is reached, the solution separates into two distinct phases, one dilute and the other considerably more concentrated. The most important feature to notice is that, as more polymer is added at a fixed temperature (Figure 3–80A), its concentration in each phase remains the same. To accommodate the increased amount of polymer present, the volume of the concentrated phase increases and the volume of the dilute phase decreases. These and other features of phase separation are relevant when considering biomolecular condensates, even though the latter are generally composed of mixtures of more complex biological polymers (proteins and RNA molecules).
Figure 3–80How phase diagrams are used to describe phase separations. (A) The effect of increasing the polymer concentration at constant temperature. At a low total concentration of a polymer (Ct), only a single dilute phase is observed. But as the polymer concentration is increased (red arrow), phase separation begins for Ct > C1, and a new concentrated phase now forms with the polymer at concentration C2 in equilibrium with a dilute phase at polymer concentration C1. As Ct is further increased, the phase with polymer concentration C2 increases in volume, while remaining in equilibrium with the polymer in the dilute phase at an unchanging concentration C1. Finally, for Ct > C2, there is only a single phase with concentration Ct. In the example illustrated, C2 is more than 10-fold greater than C1. (B) The effect of increasing temperature (T) at a constant total polymer concentration. As the temperature is raised in a solution that contains a phase-separated polymer from T1 to T2 (blue arrow), the concentration of polymer in its dilute phase (C1) increases and the concentration of polymer in its concentrated phase (C2) decreases. At a higher critical temperature C1 = C2, and the two phases become one. This occurs because the unfavorable entropy change for demixing (ΔS) makes an increasingly large, unfavorable contribution to the net free-energy change at higher temperatures (via the –TΔS term in the equation for ΔG), eventually preventing any separation of phases.
A Comparison of Three Important Types of Large Biological Assemblies
It has long been recognized that eukaryotic cells contain many membrane-enclosed compartments central to cell biology. These take the form of organelles such as the nucleus, endoplasmic reticulum, Golgi apparatus, and lysosome. Each such organelle concentrates a particular set of enzymes and substrates, thereby creating a specialized biochemistry in its interior. Those compartments will be the subject of Chapter 12, where we will also discuss biomolecular condensates in more detail. In Table 3–3, we compare the properties of the protein machines and biomolecular condensates introduced in this chapter, both with each other and with membrane-enclosed compartments.
TABLE 3–3 Macromolecular Machines Compared to Biomolecular Condensates and Membrane-enclosed Compartments
Comparison of three types of large biochemical assemblies
Macromolecular machine
Biomolecular condensate
Membrane-enclosed compartment
Properties
Fixed macromolecular composition, with a defined stoichiometry and spatial organization of constituents
Formed from a specific set of protein molecules or from protein and RNA molecules
Assembles spontaneously and can form de novo
Nevertheless, in many cases assembly is regulated to occur at specific sites, as needed
Dynamic, often liquidlike or gel-like organization, in which RNAs and low-complexity domains of proteins form specific, but transient, interactions
Readily permeable to small molecules
Larger than most macromolecular machines
Macromolecule composition is selective, but stoichiometry is usually not fixed
Can assemble de novo and be disassembled in response to changing conditions or cellular need
Creates a distinct chemical and protein environment that is maintained by active transport across the enclosing membrane
Interior contains a variable stoichiometry of macromolecules in solution, as determined by the above transport processes
Not permeable to most small molecules
Formation usually requires a preexisting membrane-enclosed compartment of a special kind, different for each compartment
Examples
SCF ubiquitin ligase
DNA replication protein machine
Ribosome
Nuclear pore
Nucleolus
Centrosome
Stress granule
Neuronal RNA transport granule
Endoplasmic reticulum
Mitochondrion
Transport vesicle
Lysosome
Many Proteins Are Controlled by Covalent Modifications That Direct Them to Specific Sites Inside the Cell
In this chapter, we have thus far described only a few ways in which proteins are post-translationally modified. A large number of other such modifications also occur, more than 200 distinct types being known. To give a sense of the variety, Table 3–4 presents a few of the modifying groups with known regulatory roles. Like the phosphate and ubiquitin additions described previously, these groups are added and then removed from proteins according to the needs of the cell.
TABLE 3–4 Some Molecules Covalently Attached to Proteins That Regulate Protein Function
Modifying group
Some prominent functions
Phosphate on Ser, Thr, or Tyr
Drives the assembly of a protein into larger complexes (see Figure 15–11)
Methyl on Lys
Helps to create distinct regions in chromatin by forming either monomethyl, dimethyl, or trimethyl lysine in histones (see Figure 4–34)
Acetyl on Lys
Helps to activate genes in chromatin by modifying histones (see Figure 4–34)
Palmityl group on Cys
This fatty acid addition drives protein association with membranes (see Figure 10–18)
N-Acetylglucosamine on Ser or Thr
Controls enzyme activity and gene expression in glucose homeostasis
Ubiquitin on Lys
Monoubiquitin addition regulates the transport of membrane proteins in vesicles (see Figure 13–59)
A polyubiquitin chain targets a protein for degradation (see Figure 3–66)
(Ubiquitin is a 76-amino-acid polypeptide; there are at least 10 other ubiquitin-related proteins in mammalian cells.)
A large number of proteins are modified on more than one amino acid side chain, with different regulatory events producing a different pattern of such modifications. A striking example is the protein p53, which plays a central part in controlling a cell’s response to adverse circumstances (see Figure 17–60). Through one of four different types of molecular additions, this protein can be modified at 20 different sites. Because an enormous number of different combinations of these 20 modifications are possible, the protein’s behavior can in principle be altered in a huge number of ways. Such modifications will often create a site on the modified protein that binds it to a scaffold protein in a specific region of the cell, thereby connecting it—via the scaffold—to the other proteins required for a reaction at that site. The effects can include moving the modified protein either into or out of a specific biomolecular condensate.
One can view each protein’s set of covalent modifications as a combinatorial regulatory code. Specific modifying groups are added to or removed from a protein in response to signals, and the code then alters protein behavior—changing the activity or stability of the protein, its binding partners, and/or its specific location within the cell (Figure 3–81). As a result, the cell is able to respond rapidly and with great versatility to changes in its condition or environment.
Figure 3–81Multisite protein modification and its effects. (A) A protein that carries a post-translational addition to more than one of its amino acid side chains can be considered to carry a combinatorial regulatory code. Multisite modifications are added to (and removed from) a protein through signaling networks, and the resulting combinatorial regulatory code on the protein is read to alter its behavior in the cell. (B) The pattern of some covalent modifications to the protein p53.
A Complex Network of Protein Interactions Underlies Cell Function
There are many challenges facing cell biologists in this information-rich era when a huge number of complete genome sequences are known. One is the need to dissect each one of the thousands of protein machines that exist in an organism such as ourselves. To understand these remarkable protein complexes, each will need to be reconstituted from its purified protein parts, so that we can study its detailed mode of operation under controlled conditions in a test tube, free from all other cell components. This alone is a massive task. But we now know that each of these subcomponents of a cell also interacts with other sets of macromolecules, creating a large network of protein–protein and protein–nucleic acid interactions throughout the cell. To understand the cell, therefore, we will need to analyze most of these other interactions as well.
We can begin to gain a sense of the nature of intracellular protein networks from a particularly well-studied example described in Chapter 16: the many dozens of proteins in the actin cytoskeleton that interact to control actin filament behavior (see Panel 16–3, p. 965). Biochemists and structural biologists are, in principle, able to purify all of these different actin-accessory proteins to study their effects on actin filaments individually and in combination, and to determine all of their protein–protein interactions and their atomic structures. But to truly understand the actin cytoskeleton will require that we also learn how to use this data to compute how any particular mixture of these components present in an individual cell creates that cell’s observed set of three-dimensional networks of actin structures—a goal that currently seems out of reach.
Of course, understanding the cell will require much more than understanding actin. In recent years, as described in Chapter 8, robotics has been harnessed to a set of powerful technologies to produce enormous protein interaction maps (Figure 3–82). The data obtained suggest that each of the roughly 10,000 different proteins in a human cell interacts with 5–10 different partners, illustrating the challenges that face scientists working to understand the complexity of cell chemistry.
Figure 3–82A network of protein-binding interactions in the cells of the fruit fly, Drosophila. Each line connecting a pair of dots (proteins) indicates a protein–protein interaction. Labels are used to denote a few of the highly interactive groups of proteins whose functions are described in this textbook. (From K.G. Guruharsha et al., Cell 147:690–703, 2011. With permission from Elsevier.)
What does the future hold? Despite the enormous progress made in recent years, we cannot yet claim to understand even the simplest known cells, such as the small Mycoplasma bacterium formed from only about 500 gene products (see Figure 1–8). How then can we hope to understand a human? Clearly, a great deal of new biochemistry will be essential, in which each protein in a particular interacting set is purified so that its chemistry and interactions can be dissected in a test tube. But in addition, more powerful ways of analyzing networks will be needed using mathematical and computational tools not yet invented. Clearly, there are many wonderful challenges that remain for future generations of cell biologists.
Protein Structures Can Be Predicted and New Proteins Designed
Because the structures and functions of proteins are encoded in their amino acid sequences, in principle it is possible to predict the structures and functions of proteins directly from their amino acid sequences. We should also be able to create proteins with entirely new structures and functions by designing new amino acid sequences to produce these structures and functions, encoding them in synthetic genes. Success in the first endeavor would transform our ability to understand how the biology of an organism is encoded in the DNA sequence of its genome. Success in the second endeavor could lead to a new generation of designed proteins that address some of the twenty-first-century challenges confronting humanity.
There are major challenges in both of the above areas. A first challenge is the very large number of potential structures that are possible for any given amino acid sequence. Because, as we have seen, a protein folds to its lowest free-energy state, one needs to use physics to compute the energy of each protein conformation. But the number of possible conformations for even a relatively short protein of 100 amino acids is of the order 3100, as each amino acid has on average 3 or more rotatable bonds. Success in predicting protein structure and in designing new proteins thus requires computational methods for very efficient searching through huge numbers of structures.
Progress has been made in recent years. For small proteins or for proteins from very large families to help constrain the problem, large-scale computer searches for the lowest energy state can often accurately predict protein structure starting from amino acid sequence. Recently developed deep learning approaches using artificial intelligence (AI) can produce even more accurate protein structure predictions. Conversely, many new protein structures and functions have been created from scratch by designing new sequences in which the lowest energy state has the desired structure and function (Figure 3–83).
Figure 3–83Some examples of designed proteins. The amino acid sequences of the three proteins illustrated were determined computationally, being selected so that each protein would adopt a specifically designed three-dimensional conformation in its lowest energy state. After each protein was produced in a bacterium using genetic engineering techniques, its actual structure was then determined and shown to be the same as that intended by the designer. (A) A small protein of 122 amino acids. (B) A protein that creates an octahedral shell formed from 24 identical subunits, only 8 of which are shown. (C) A protein that consists of an antiparallel three-helix bundle. (A, PDB code 2N76; B, PDB code 3VCD; C, PDB code 4TQL).
While this progress suggests that the protein-folding problem is not intractable, huge challenges remain. Predicting function from structure is even more difficult: while in some cases function can be predicted from structure by analogy to other proteins with similar structures and already known functions, this can be problematic because even a few amino acid changes can considerably change function; for example, the identity of the substrate that an enzyme acts upon. On the design side, while it has been possible to design new proteins with new structures and binding activities, it remains a big challenge to match the remarkable activities of natural enzymes and the sophisticated information integration and force generation of natural molecular machines.
Summary
The function of a protein largely depends on the detailed chemical properties of its surface. Enzymes are catalytic proteins that greatly accelerate the rates of covalent bond making and breaking. They do this by binding the high-energy transition state for a specific reaction path, lowering that reaction’s activation energy. The rates of enzyme-catalyzed reactions are often so fast that they are limited only by diffusion.
Proteins can reversibly change their shape when ligands bind to their surface. The allosteric changes in protein conformation produced by one ligand affect the binding of a second ligand, and this linkage between two ligand-binding sites provides a crucial mechanism for regulating cell processes. Metabolic pathways, for example, are controlled by feedback regulation: some small molecules inhibit and other small molecules activate enzymes early in a pathway. Enzymes controlled in this way generally form symmetrical assemblies, allowing cooperative conformational changes to create a steep response to changes in the concentrations of the ligands that regulate them.
The expenditure of chemical energy can drive unidirectional changes in protein shape. By coupling allosteric shape changes to the hydrolysis of a tightly bound ATP molecule, for example, proteins can do useful work, such as generating a mechanical force or moving for long distances in a single direction. The three-dimensional structures of proteins have revealed how a small local change caused by nucleoside triphosphate hydrolysis is amplified to create major changes elsewhere in the protein. Highly efficient protein machines are formed by incorporating many different protein molecules into larger assemblies that coordinate the allosteric movements of the individual components. Machines of this type perform most of the important reactions in cells. They and other specific macromolecules can be brought together in large, liquid-like assemblies known as biomolecular condensates, which are created by weak, fluctuating interactions between multivalent protein and RNA scaffolds.
Proteins are subjected to many reversible, post-translational modifications, such as the covalent addition of a phosphate or an acetyl group to a specific amino acid side chain. The addition of these modifying groups is used to regulate the activity of a protein, changing its conformation, its binding to other proteins, and its location inside the cell. A typical protein in a cell will interact with more than five different partners. Understanding the large protein networks inside cells will require biochemistry, through which small sets of interacting proteins can be purified and their chemistry dissected in detail. In addition, new computational approaches will be required to make sense of the enormous complexity of these networks.
Protein secreted by activated B cells in response to a pathogen or foreign molecule. Binds tightly to the pathogen or foreign molecule, inactivating it or marking it for destruction by phagocytosis or complement-induced lysis.
Protein secreted by activated B cells in response to a pathogen or foreign molecule. Binds tightly to the pathogen or foreign molecule, inactivating it or marking it for destruction by phagocytosis or complement-induced lysis.
Any molecule that can bind specifically to an antibody or B cell receptor, or any protein fragment bound to an MHC protein that can bind specifically to a T cell receptor.
State in a chemical reaction where there is no net change in free energy to drive the reaction in either direction. The ratio of product to substrate reaches a constant value at chemical equilibrium.
The ratio of forward and reverse rate constants for a reaction. Equal to the association or affinity constant (Ka) for a simple binding reaction (A + B → AB). See alsoaffinity constant, association constant, dissociation constant.
Structure that forms transiently in the course of a chemical reaction and has the highest free energy of any reaction intermediate. Its formation is a rate-limiting step in the reaction.
Small molecule tightly associated with an enzyme that participates in the reaction that the enzyme catalyzes, often by forming a covalent bond to the substrate. Examples include biotin, NAD+, and coenzyme A.
(adjective allosteric) Change in a protein’s conformation brought about by the binding of a regulatory ligand (at a site other than the protein’s catalytic site) or by covalent modification. The change in conformation alters the activity of the protein; it can also form the basis of directed movement.
In ligand binding, the conformational coupling between two separate ligand-binding sites on a protein, such that a conformational change in the protein induced by binding of one ligand affects the binding of a second ligand.
A protein that can adopt at least two distinct conformations, and for which the binding of a ligand at one site causes a conformational change that alters the activity of the protein at a second site; this allows one type of molecule in a cell to alter the fate of a molecule of another type, a feature widely exploited in enzyme regulation.
An enzyme that converts GTP to GDP. GTPases fall into two large families. Large G proteins (heterotrimeric G proteins) are composed of three different subunits and mainly couple GPCRs to enzymes or ion channels in the plasma membrane. Small monomeric GTP-binding proteins (also called monomeric GTPases) consist of a single subunit and help relay signals from many types of cell-surface receptors and have roles in intracellular signaling pathways, regulating intracellular vesicle trafficking, and signaling to the cytoskeleton. Both heterotrimeric G proteins and monomeric GTPases cycle between an active GTP-bound form and an inactive GDP-bound form and frequently act as molecular switches in intracellular signaling pathways.
Monomeric GTPase of the Ras superfamily that helps to relay signals from cell-surface receptor tyrosine kinase receptors to the nucleus, frequently in response to signals that stimulate cell division. Named for the Ras gene, first identified in viruses that cause rat sarcomas.
Small, highly conserved protein present in all eukaryotic cells that can be covalently attached to lysines of other proteins. Attachment of a short chain of ubiquitins to such a lysine can tag a protein for intracellular proteolytic destruction by a proteasome.
Any one of a large number of enzymes that attach ubiquitin to a protein, often marking it for destruction in a proteasome. The process catalyzed by a ubiquitin ligase is called ubiquitylation.
An aggregate inside cells, formed by a process analogous to liquid–liquid phase separation and based on fluctuating weak interactions between scaffold proteins; concentrates selected protein and RNA molecules in a membraneless compartment.
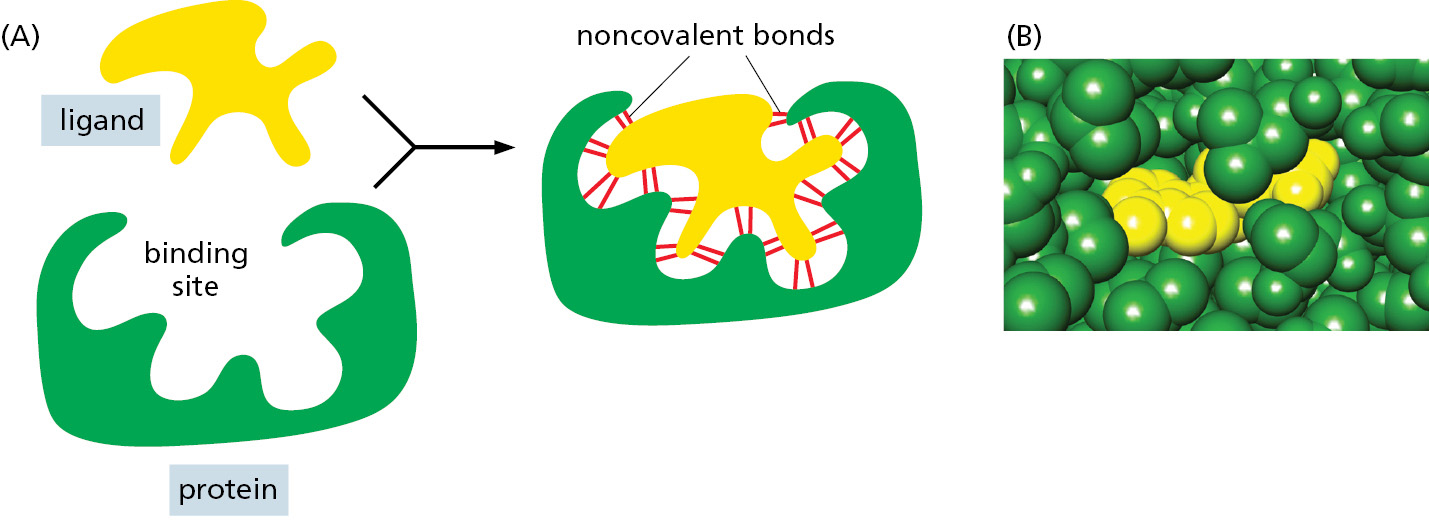
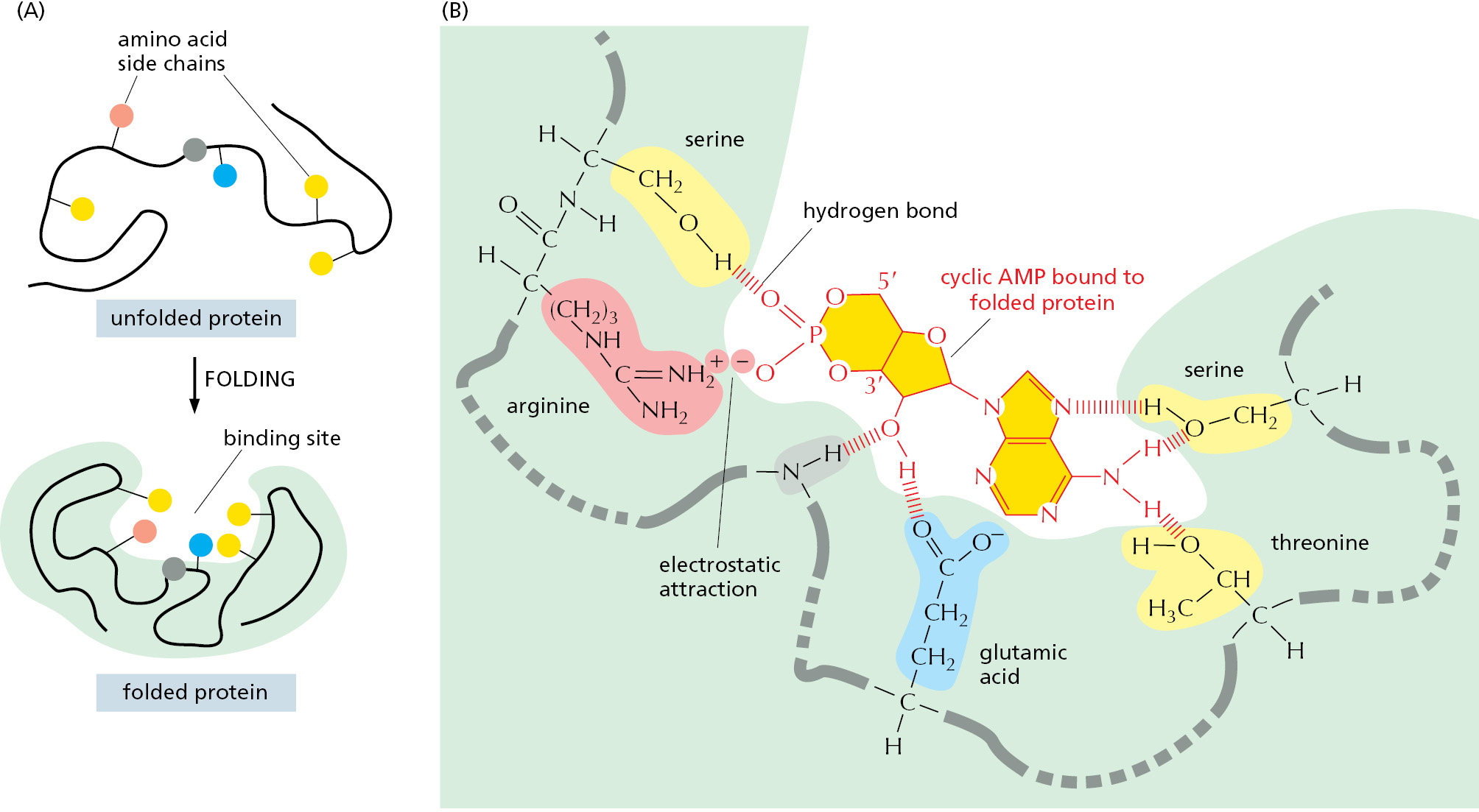
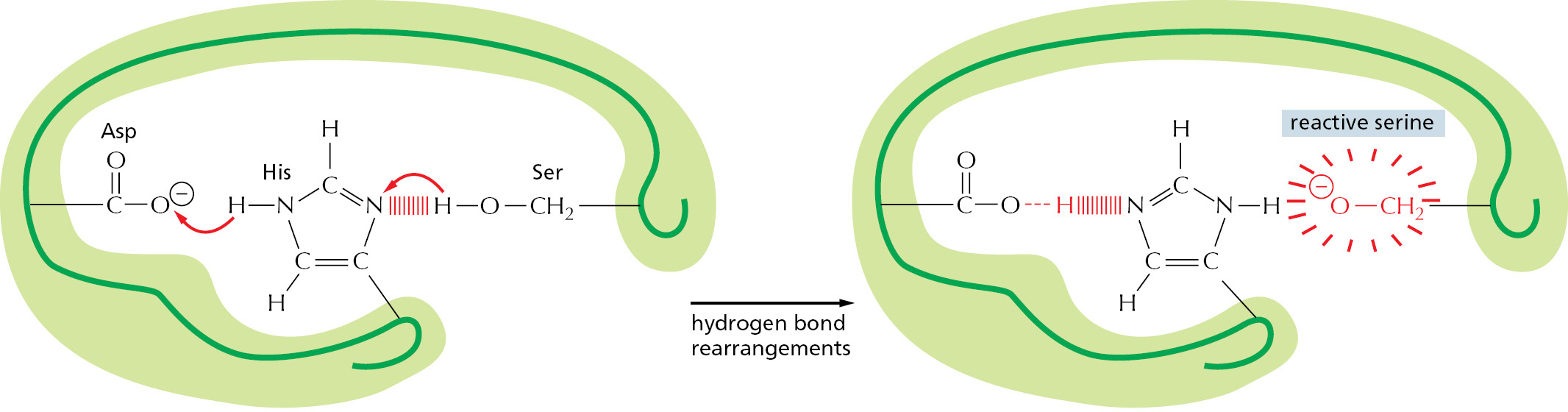
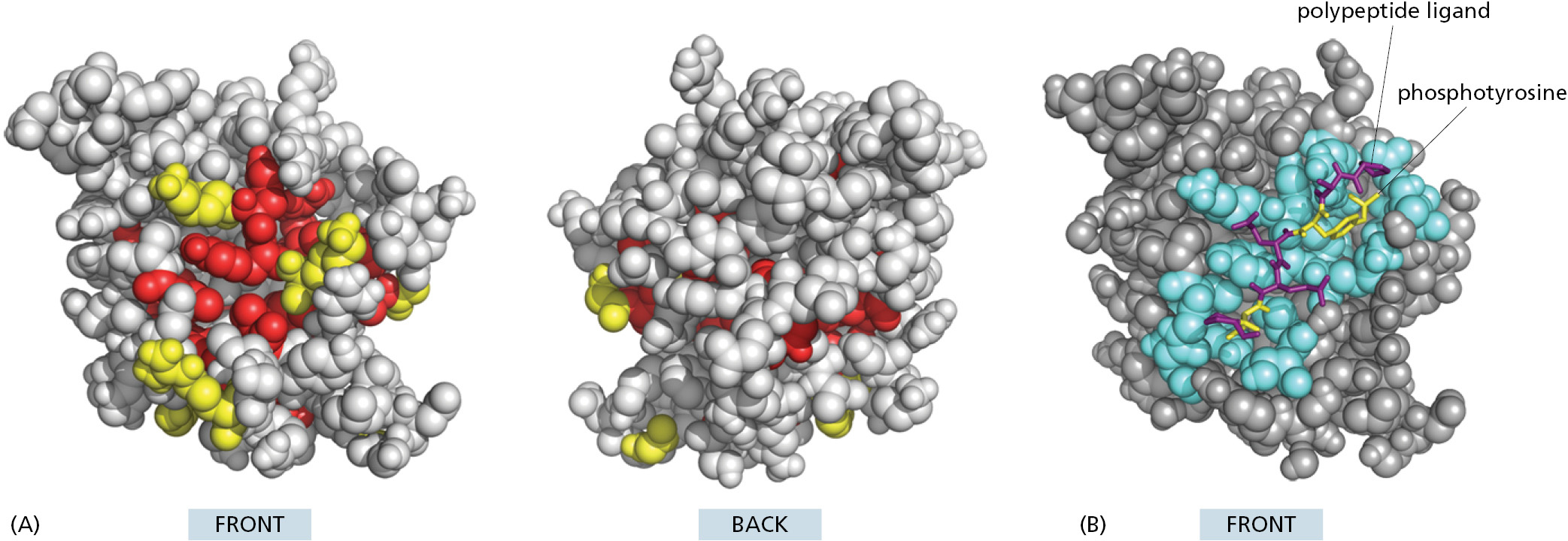
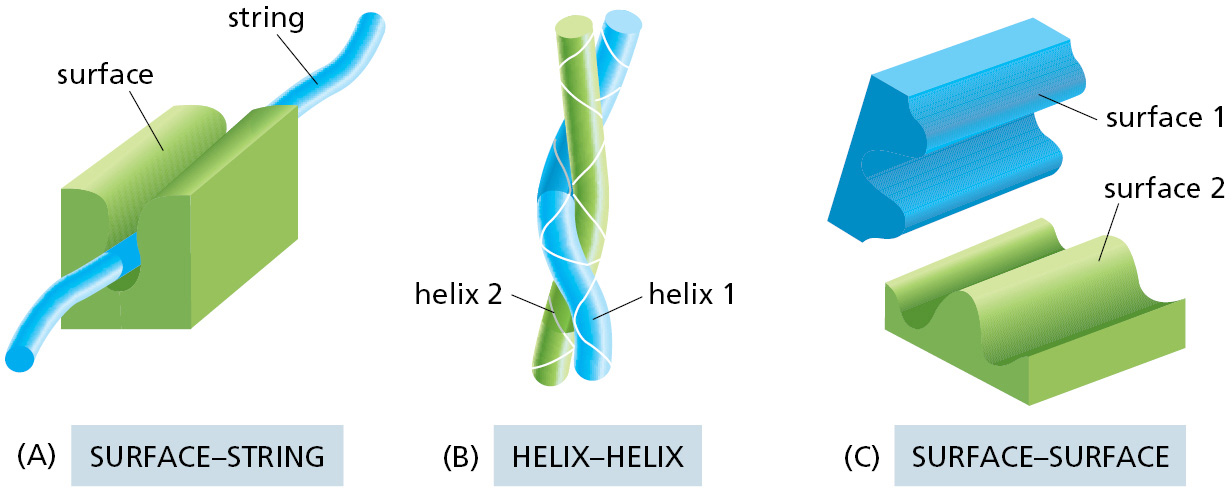
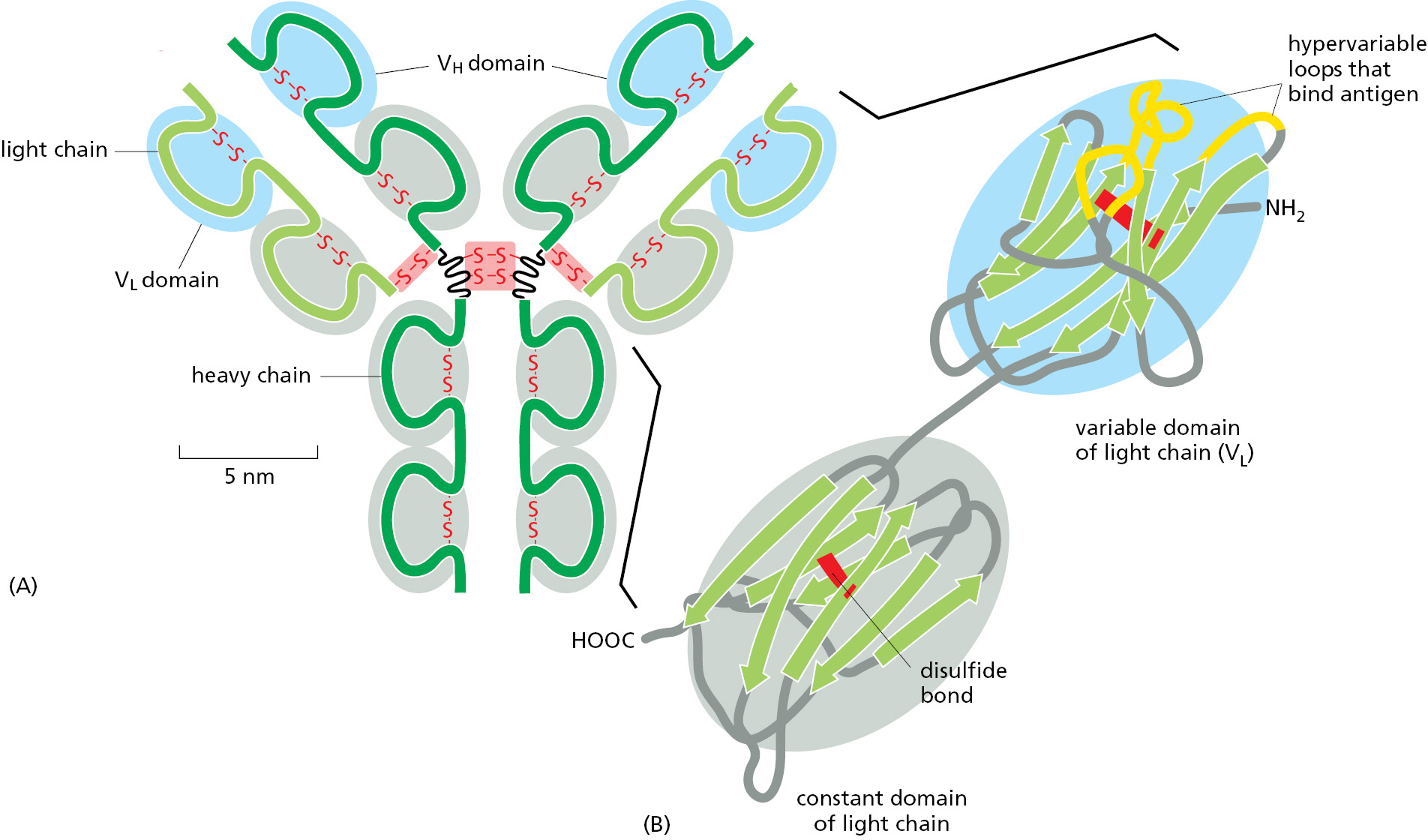
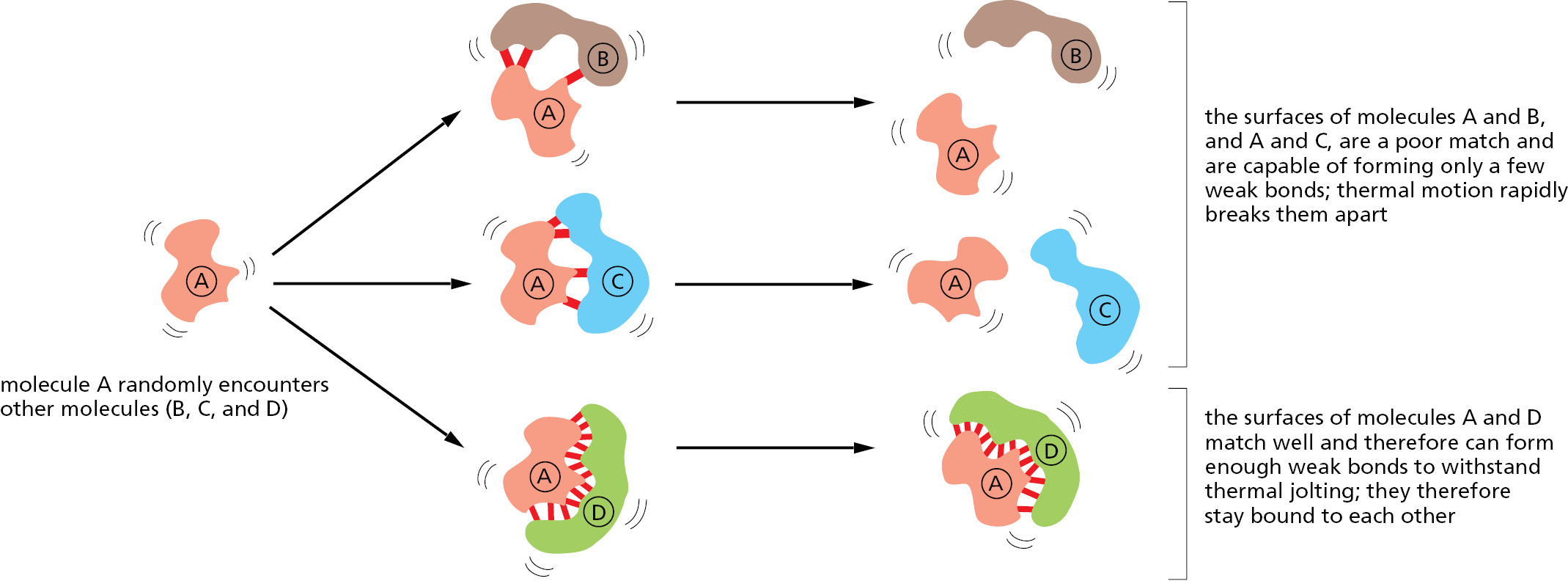
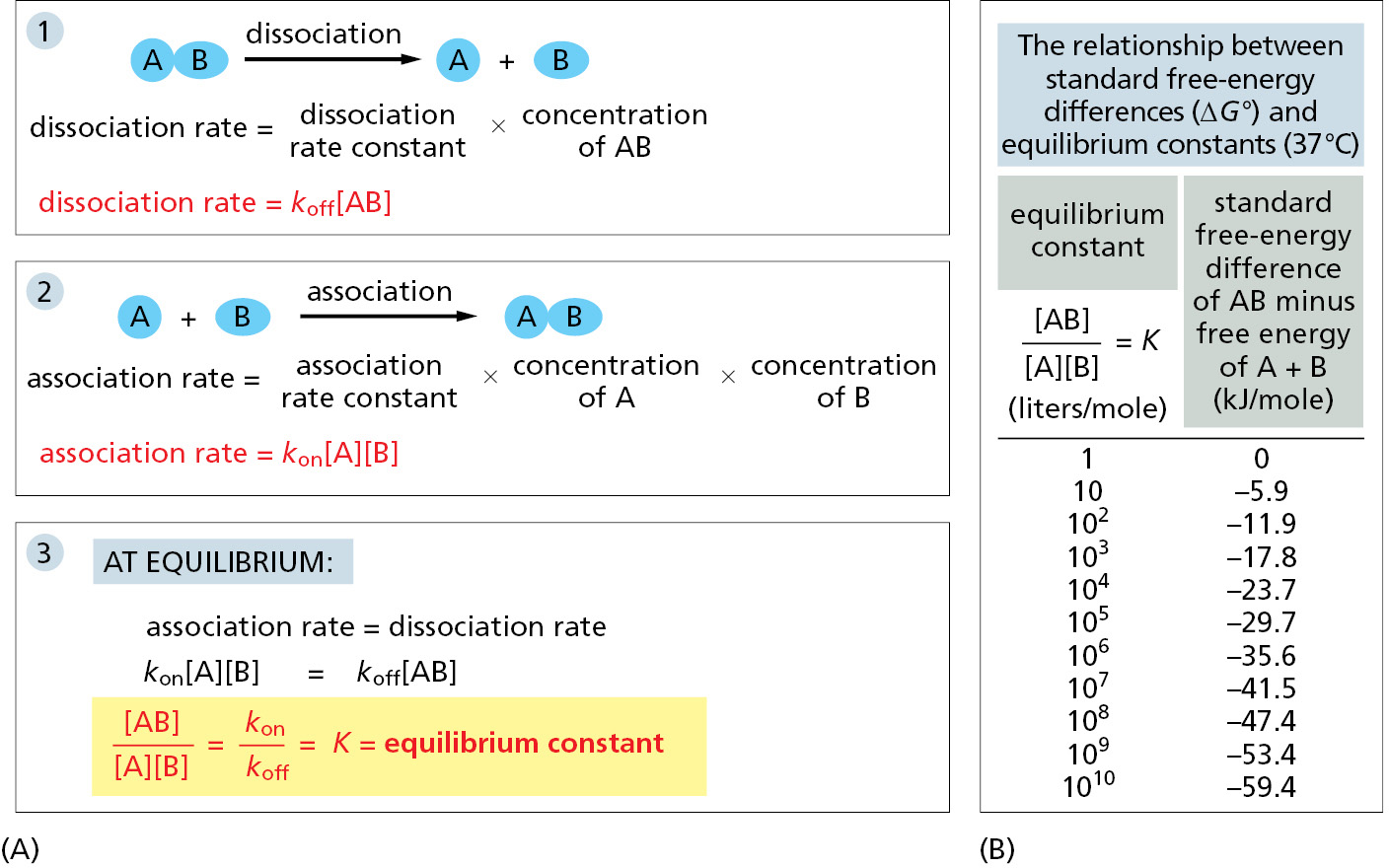

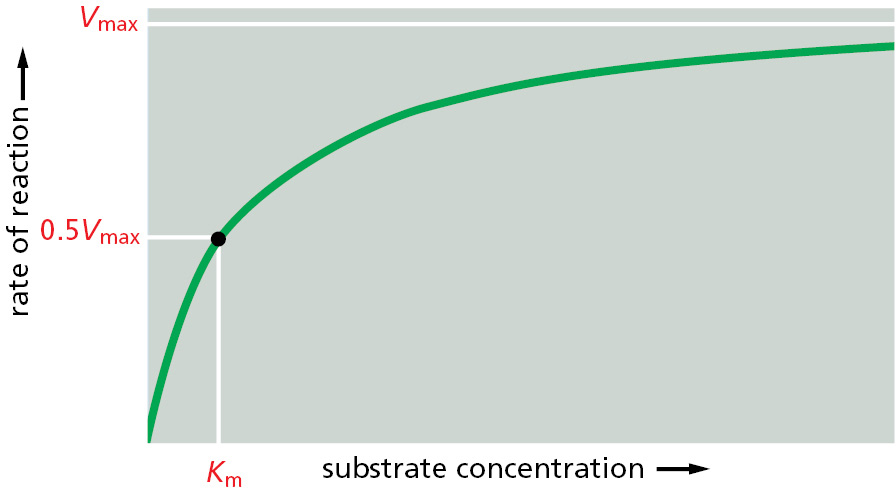
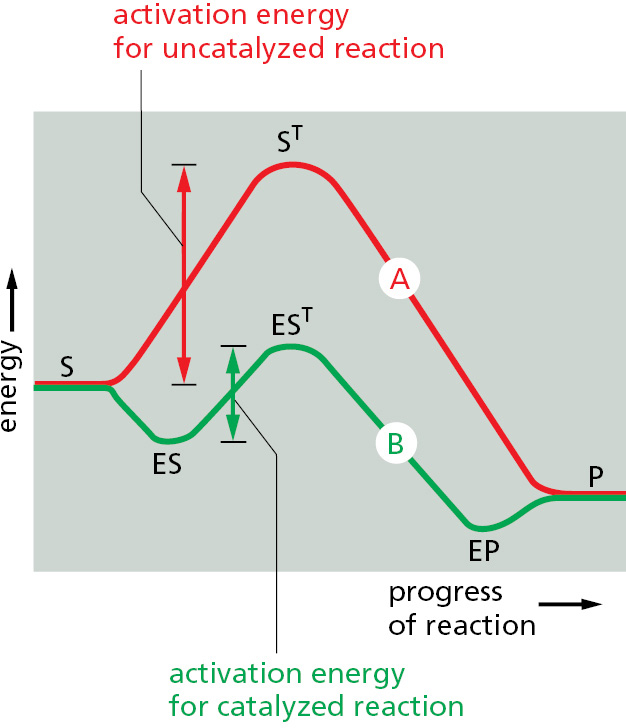
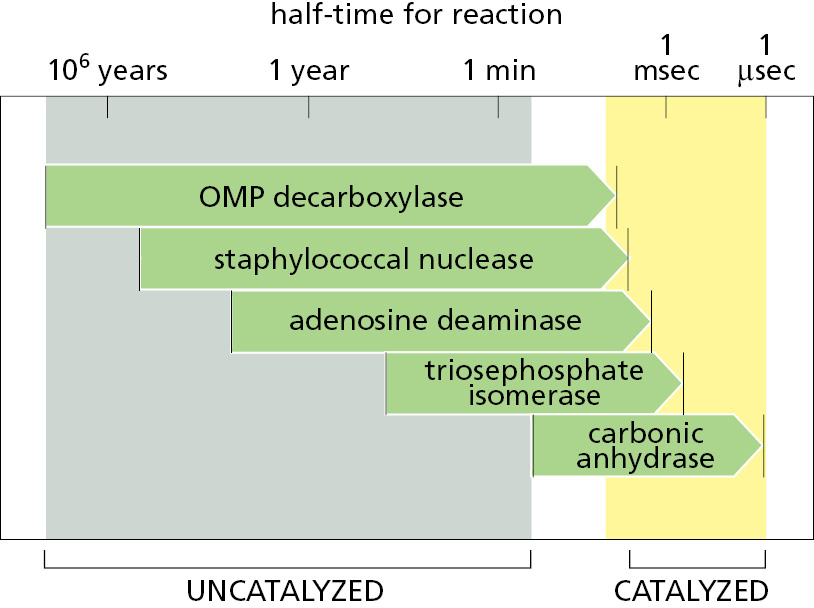
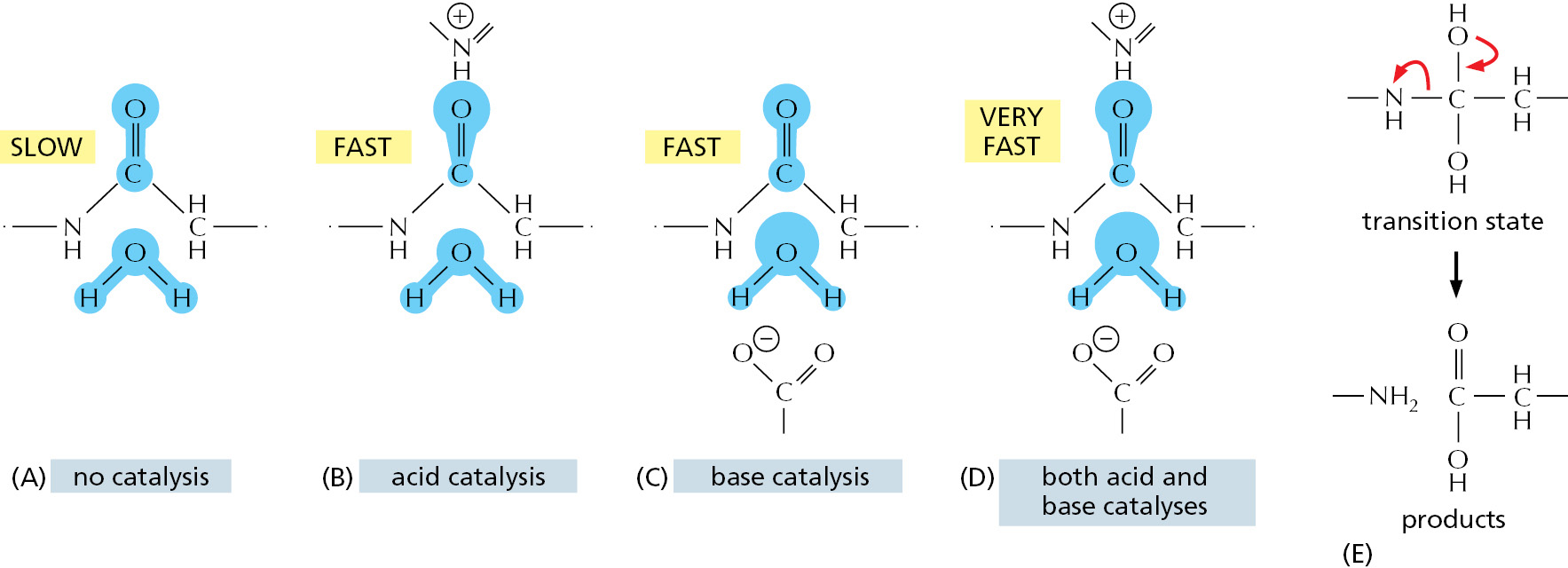

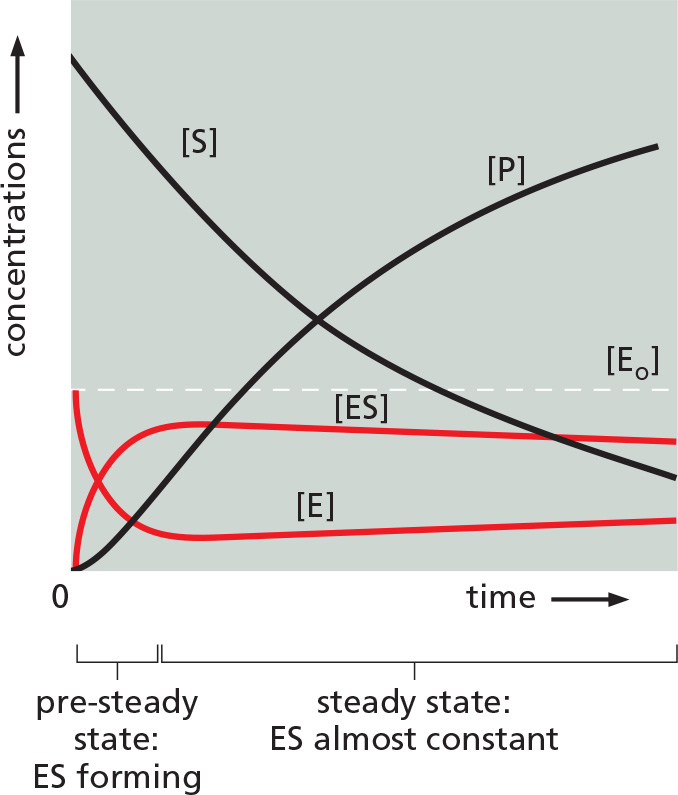

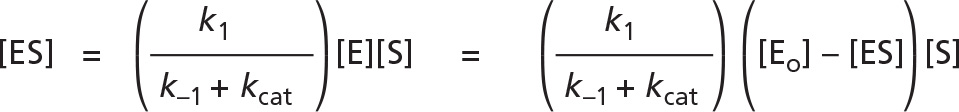
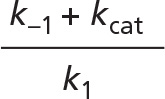
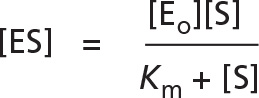
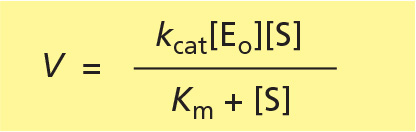
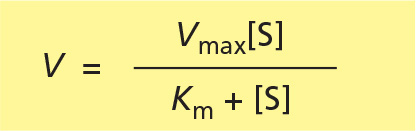
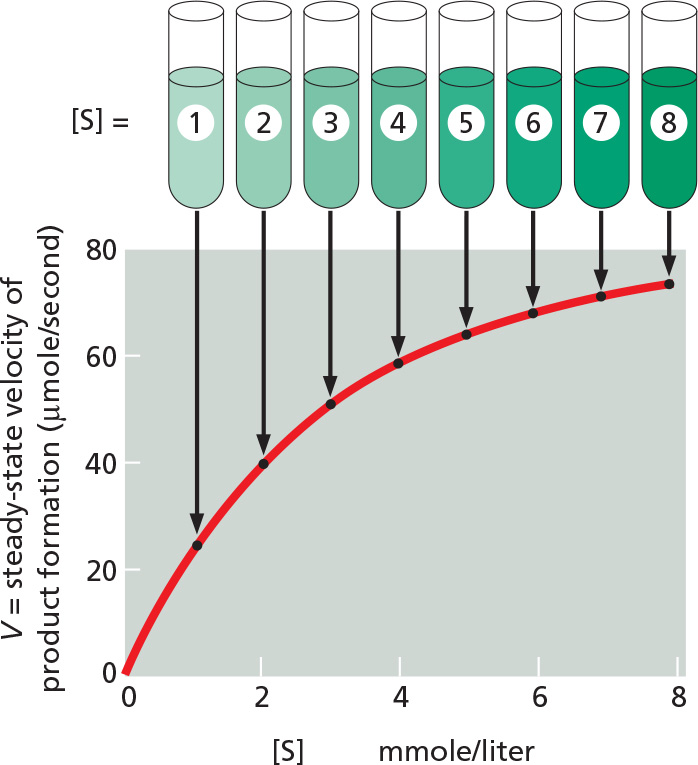
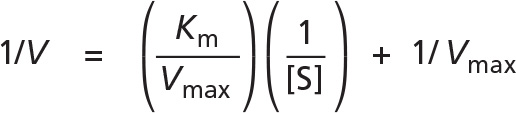
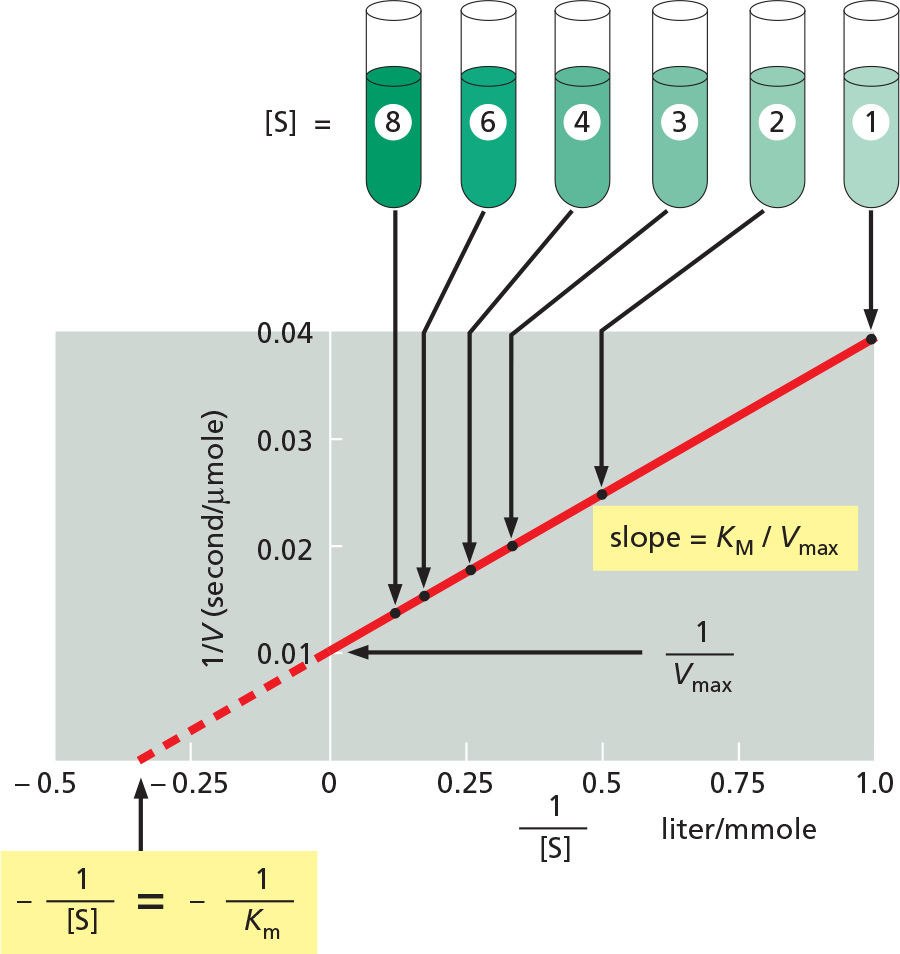
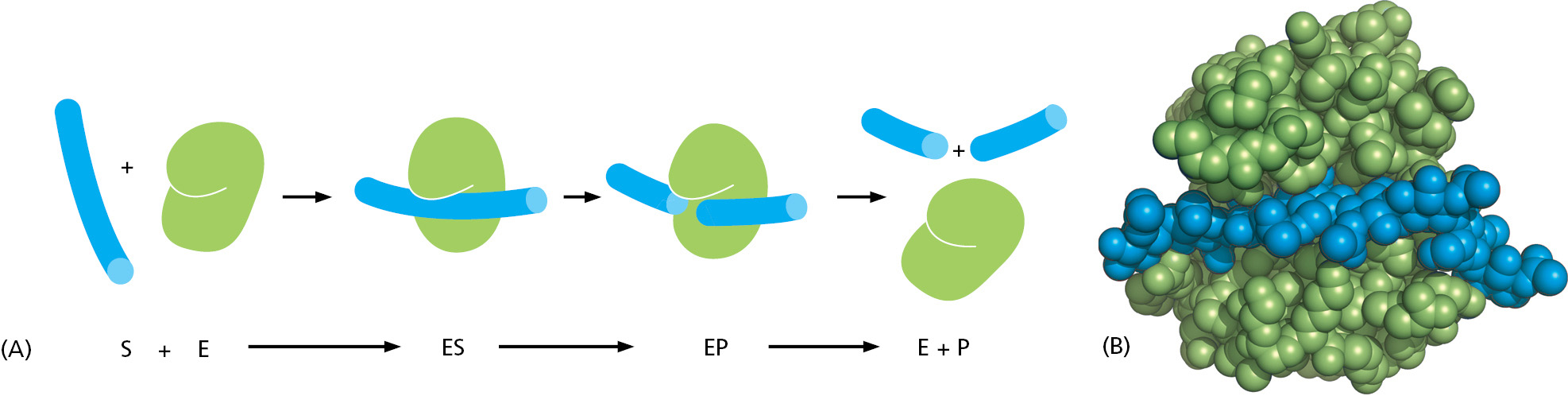
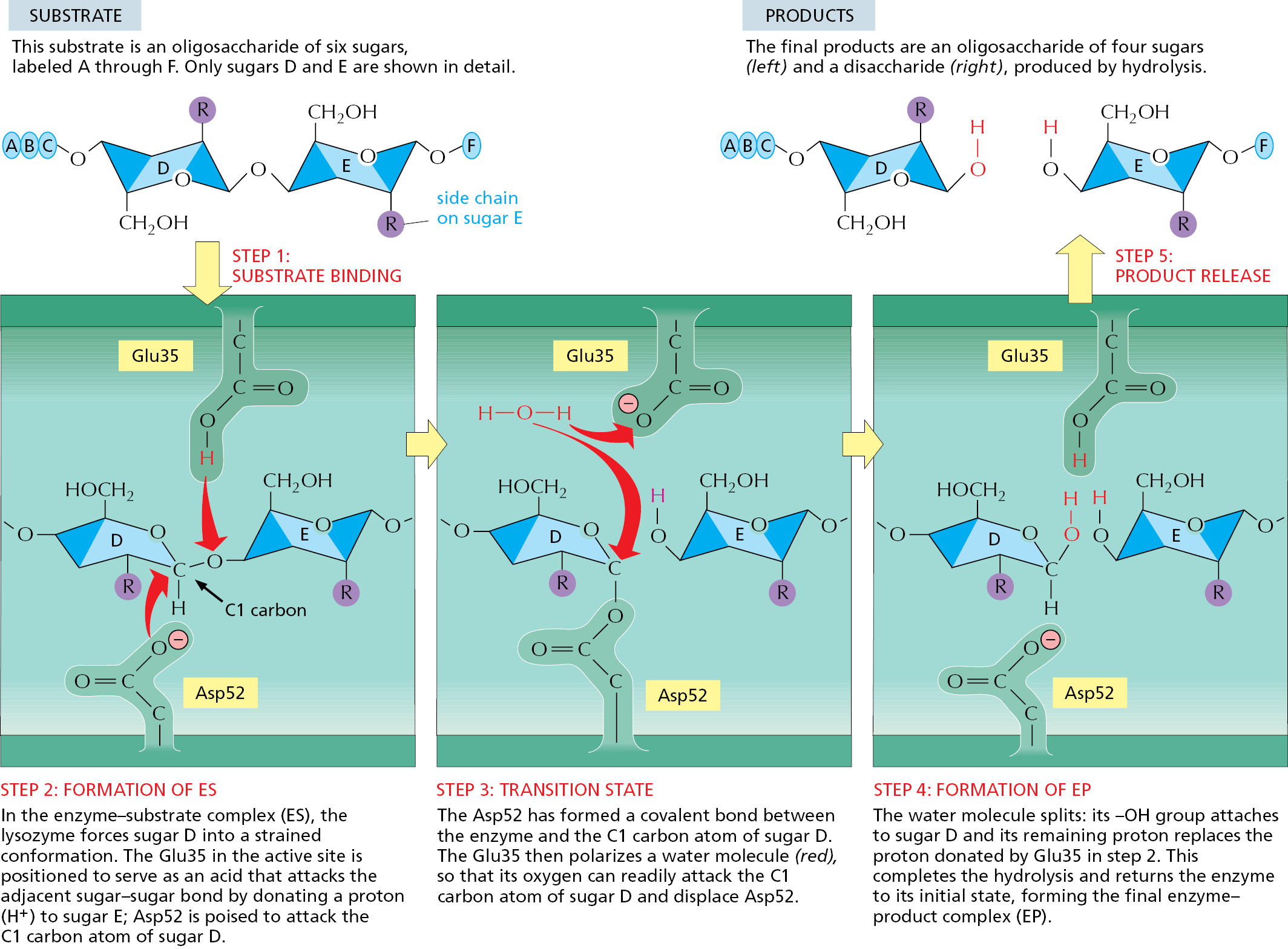
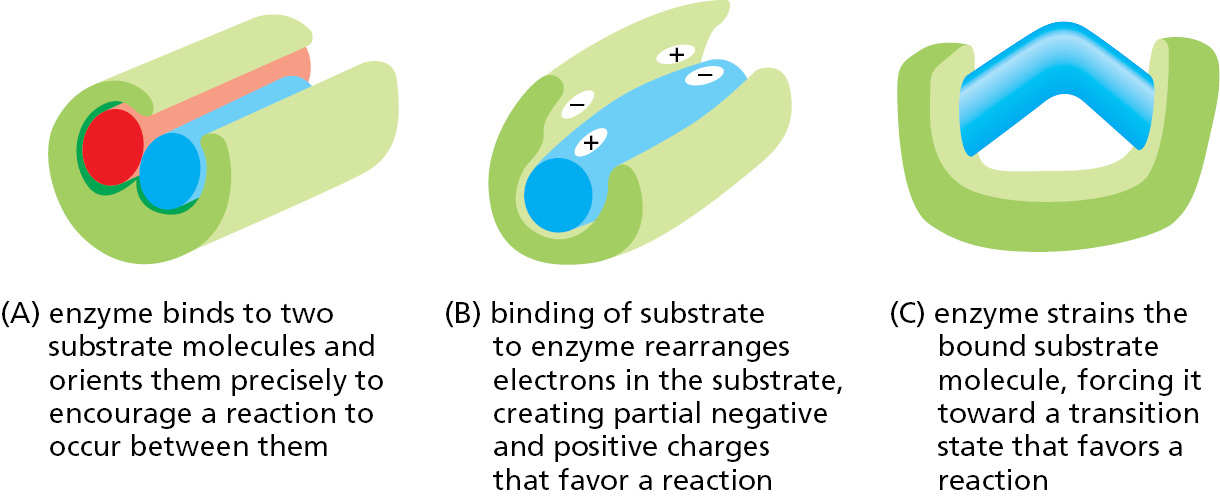
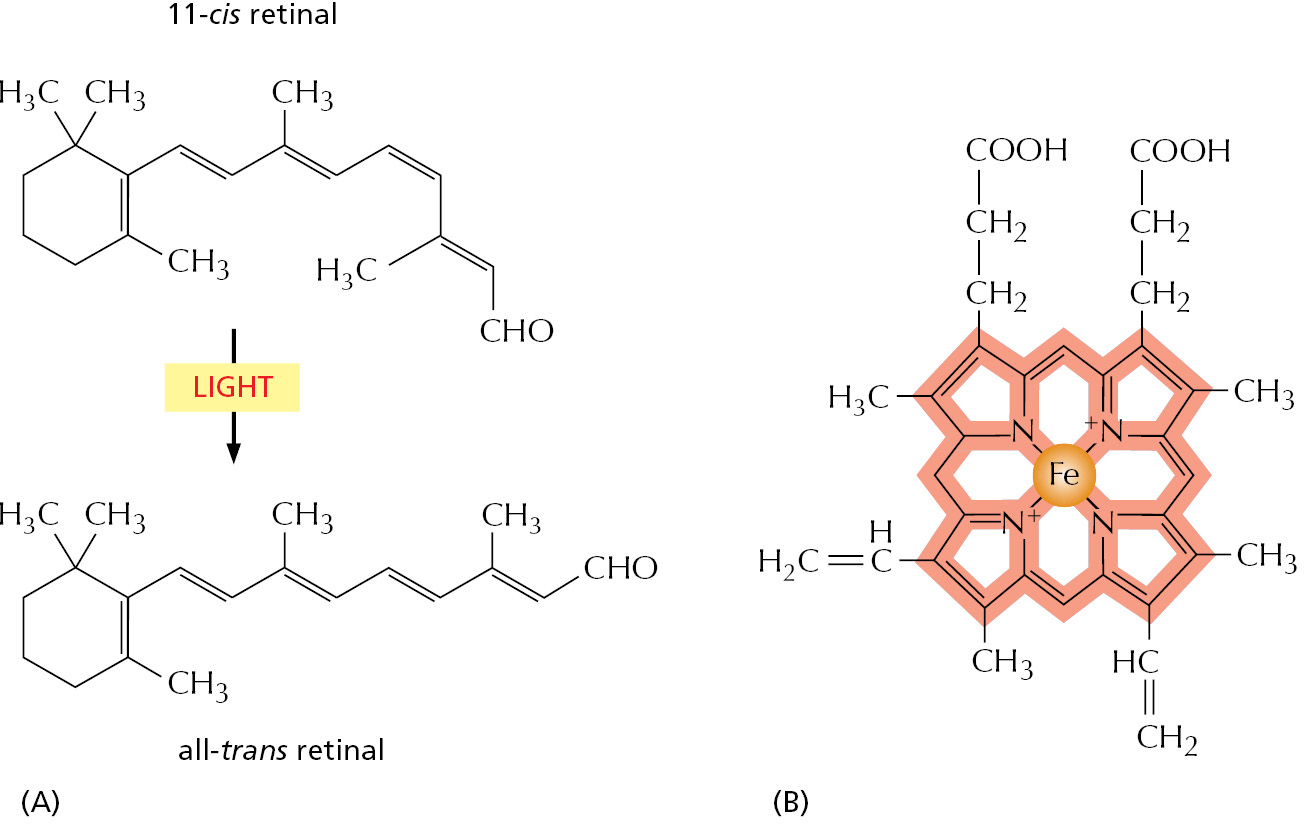
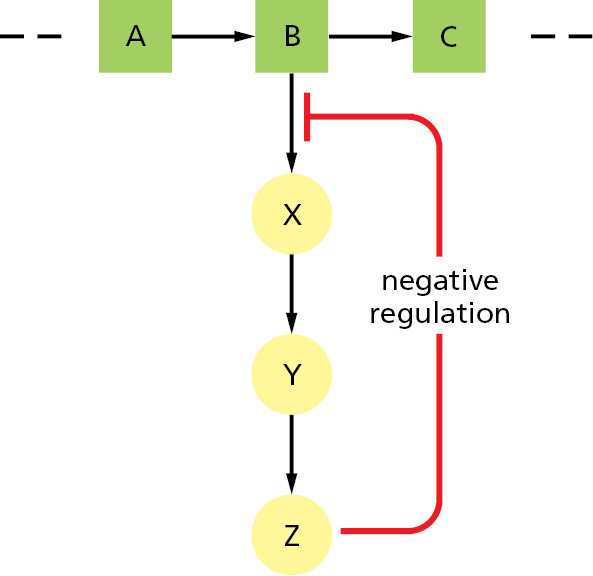
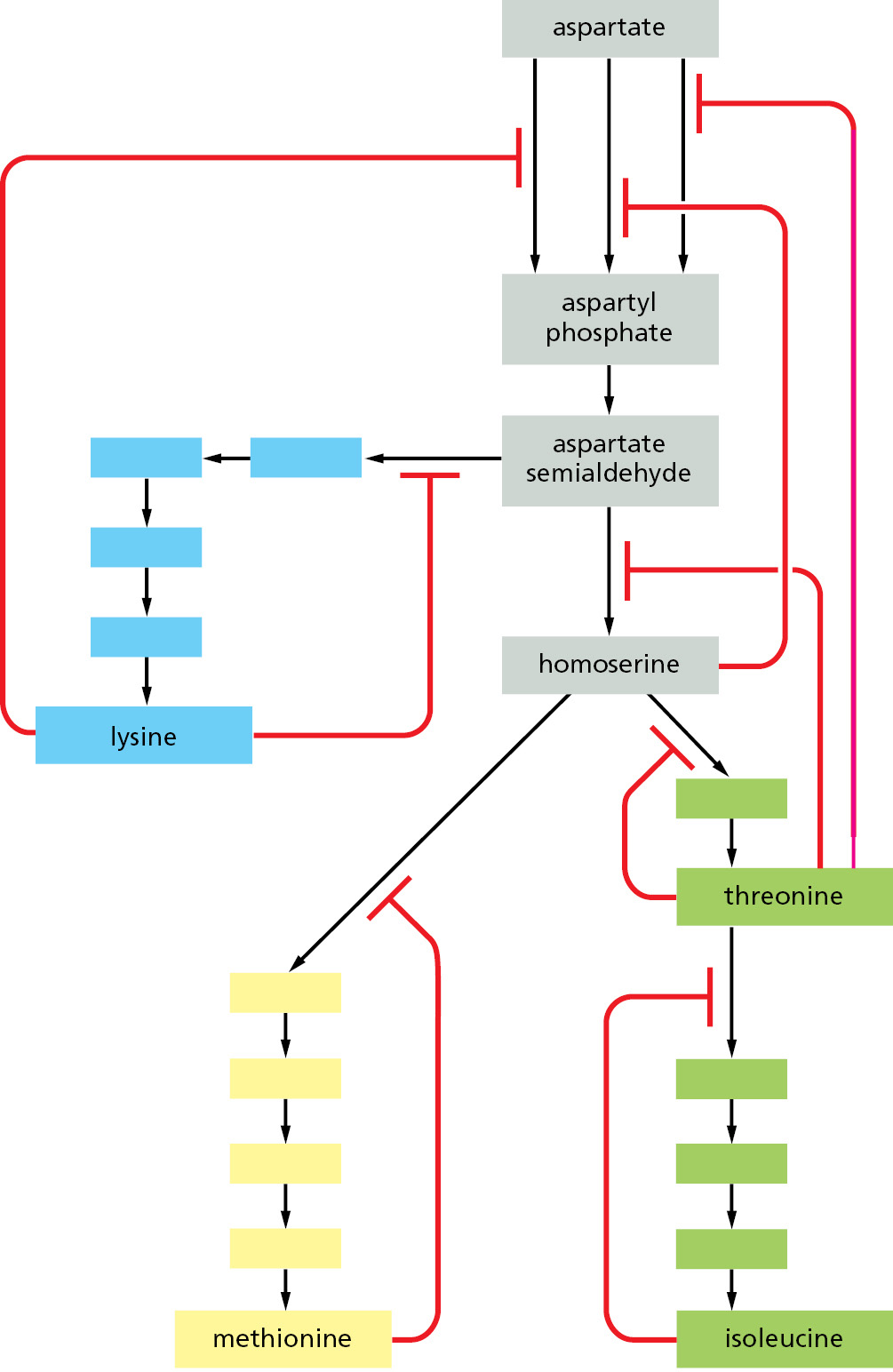
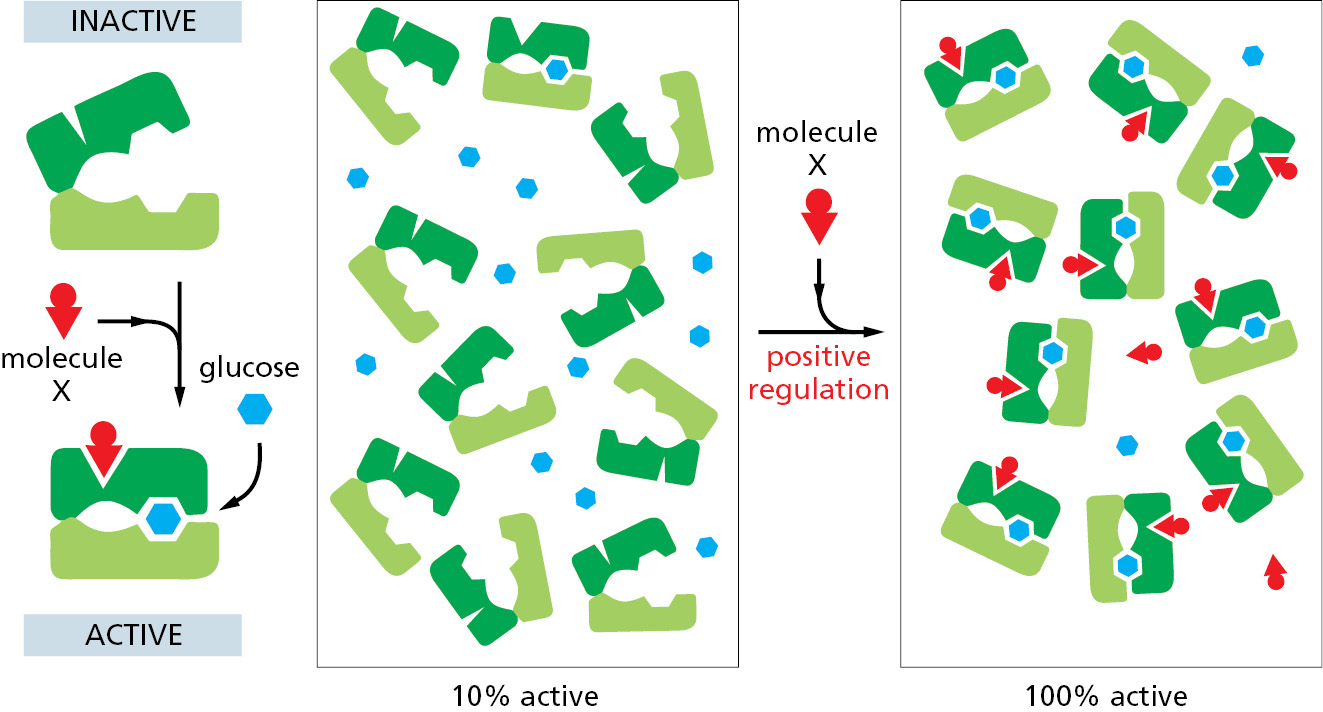
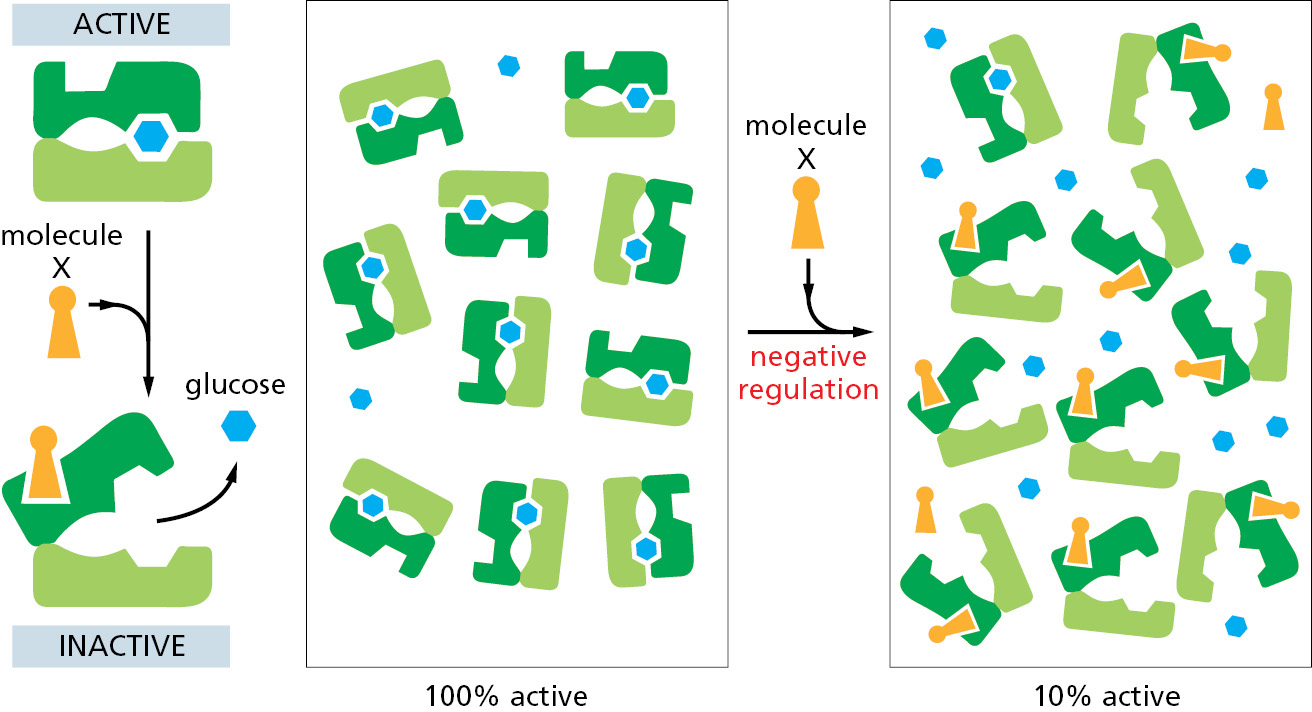
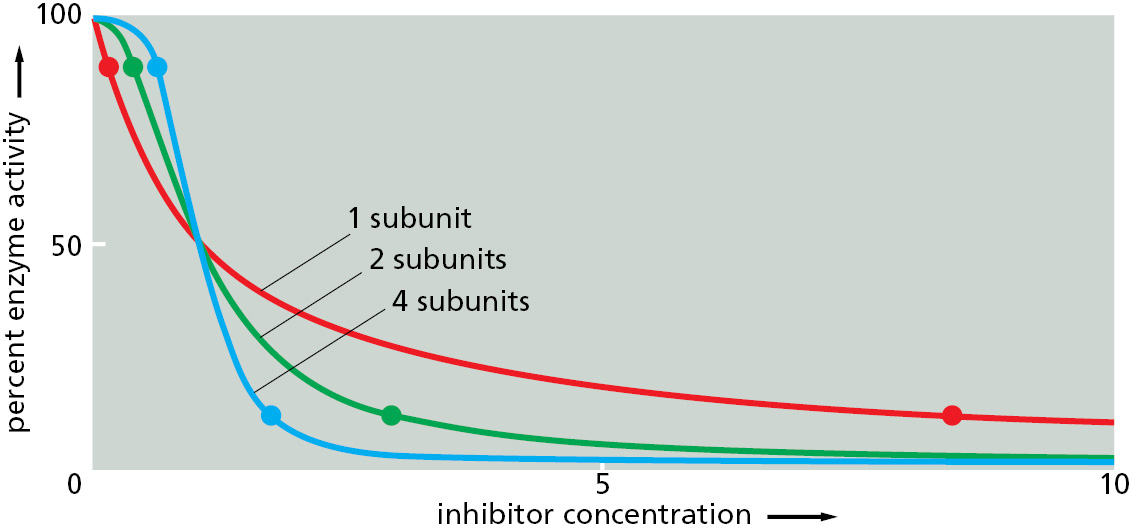
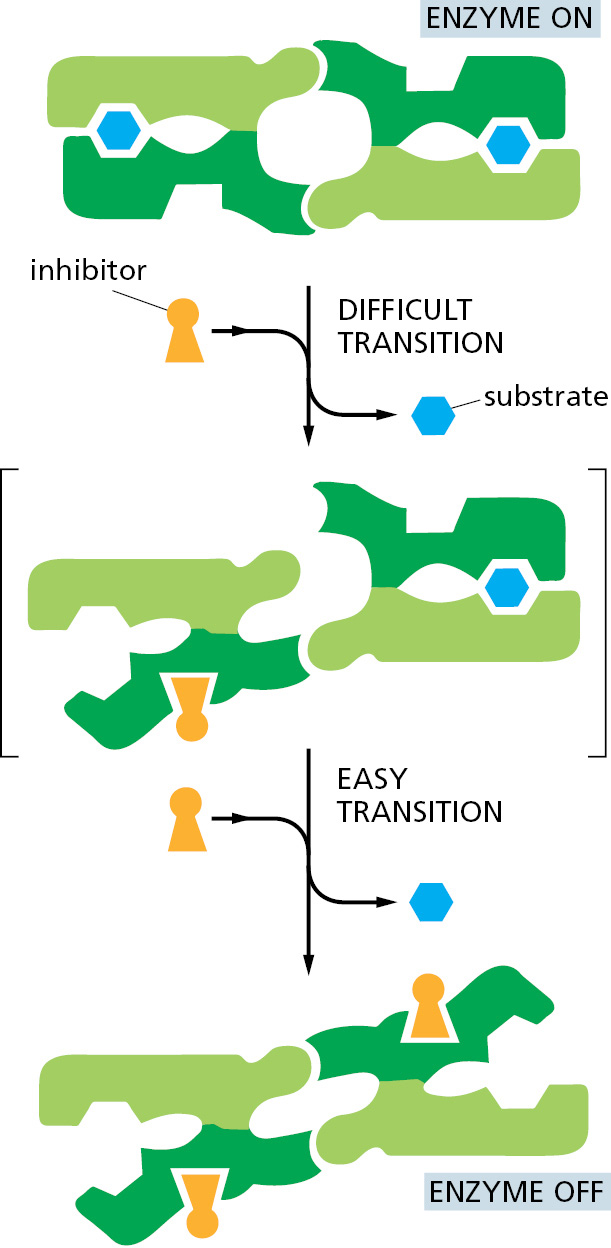
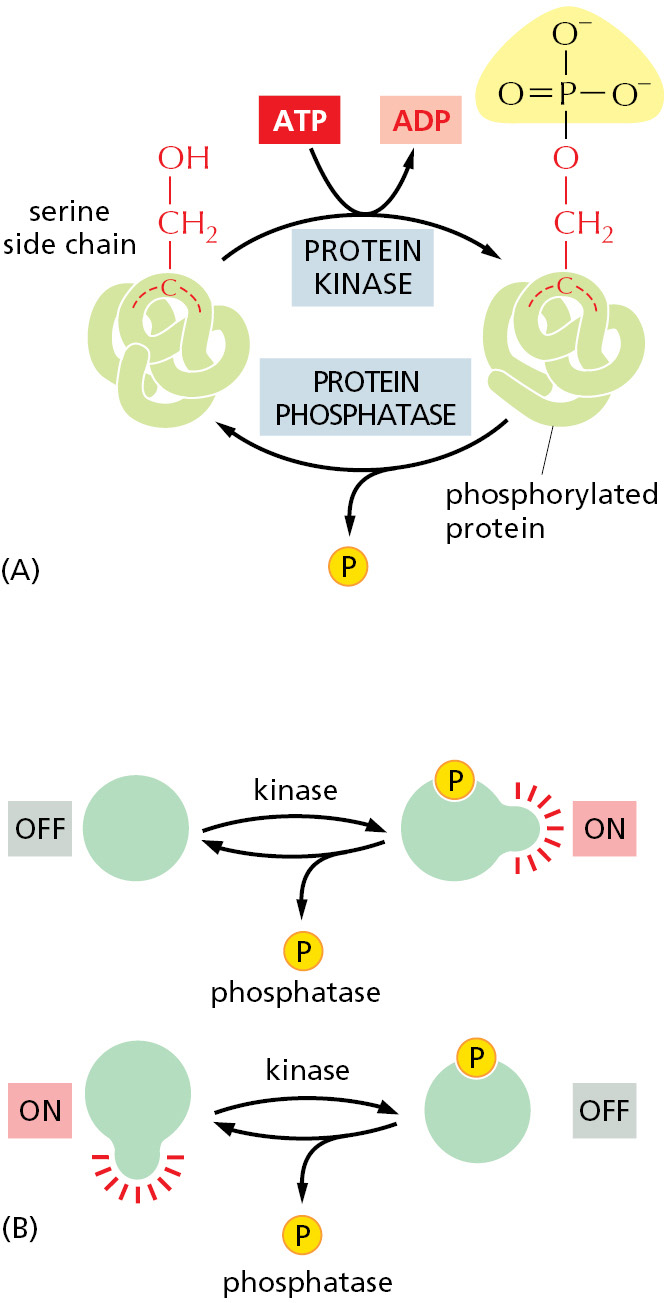
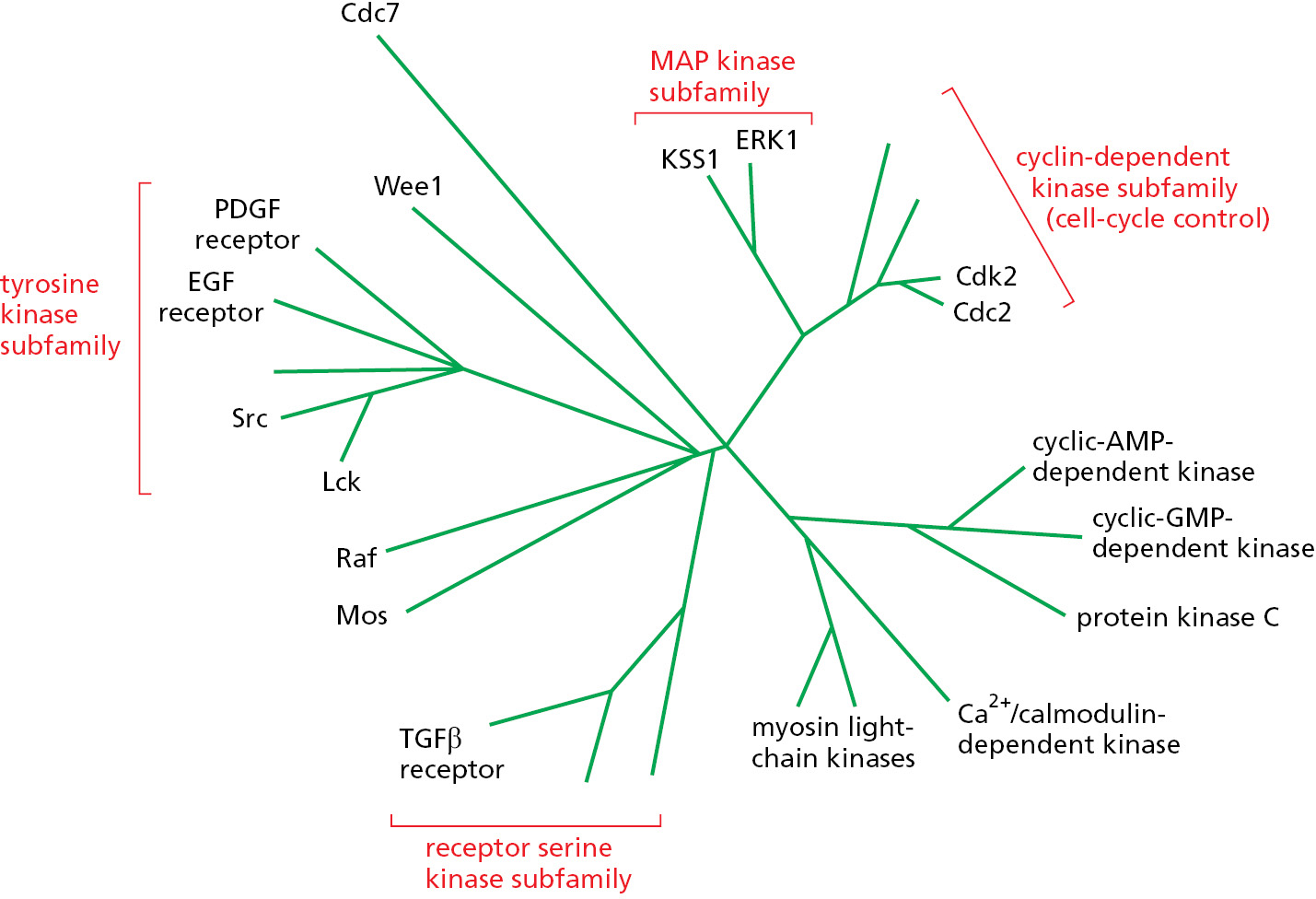
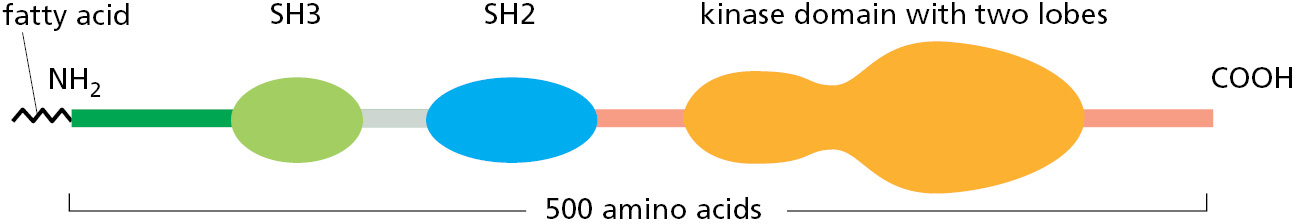
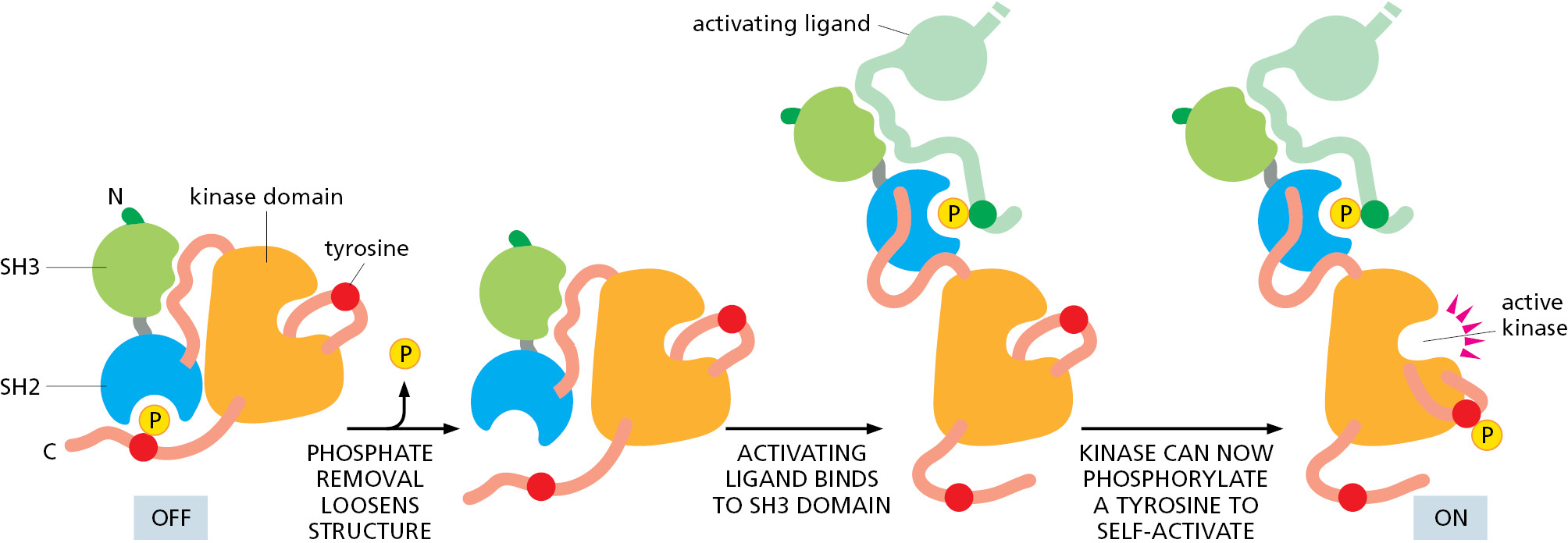
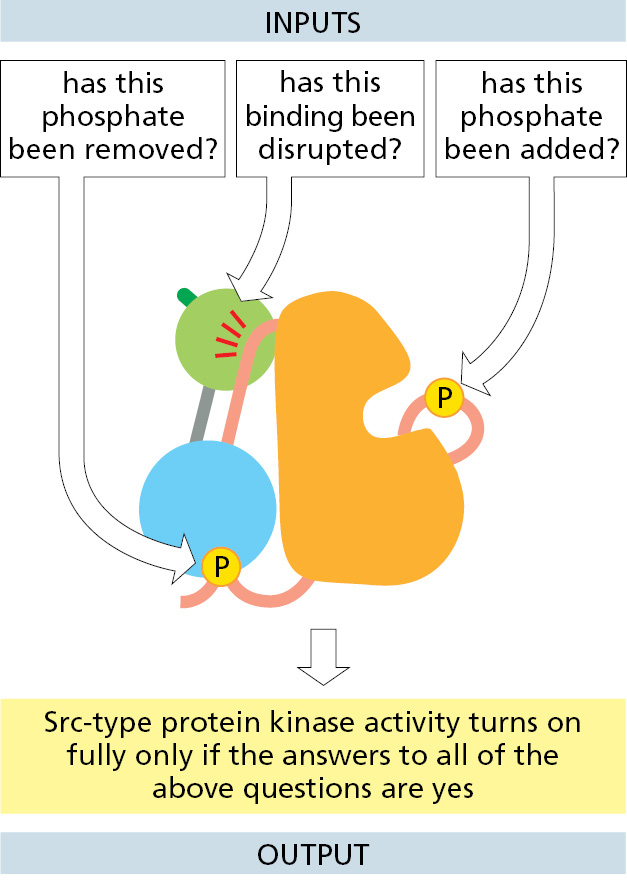
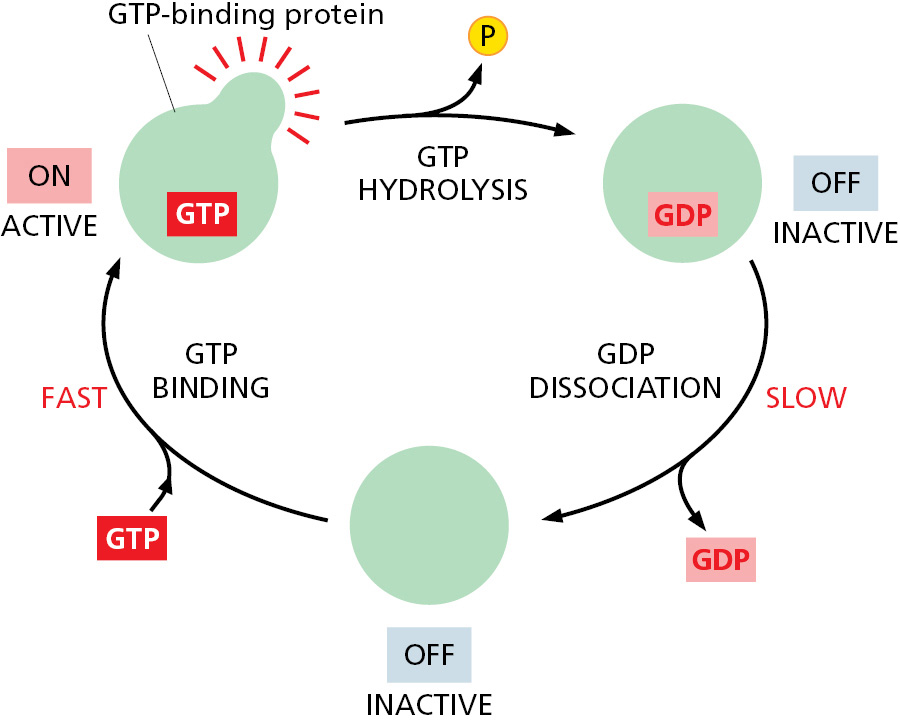
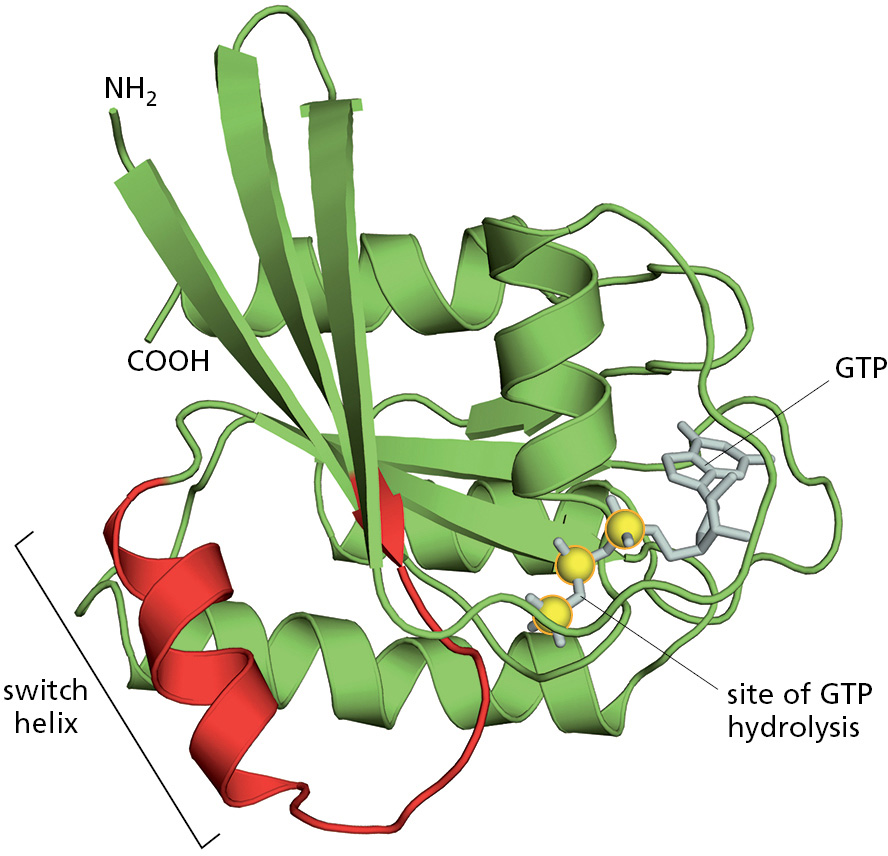
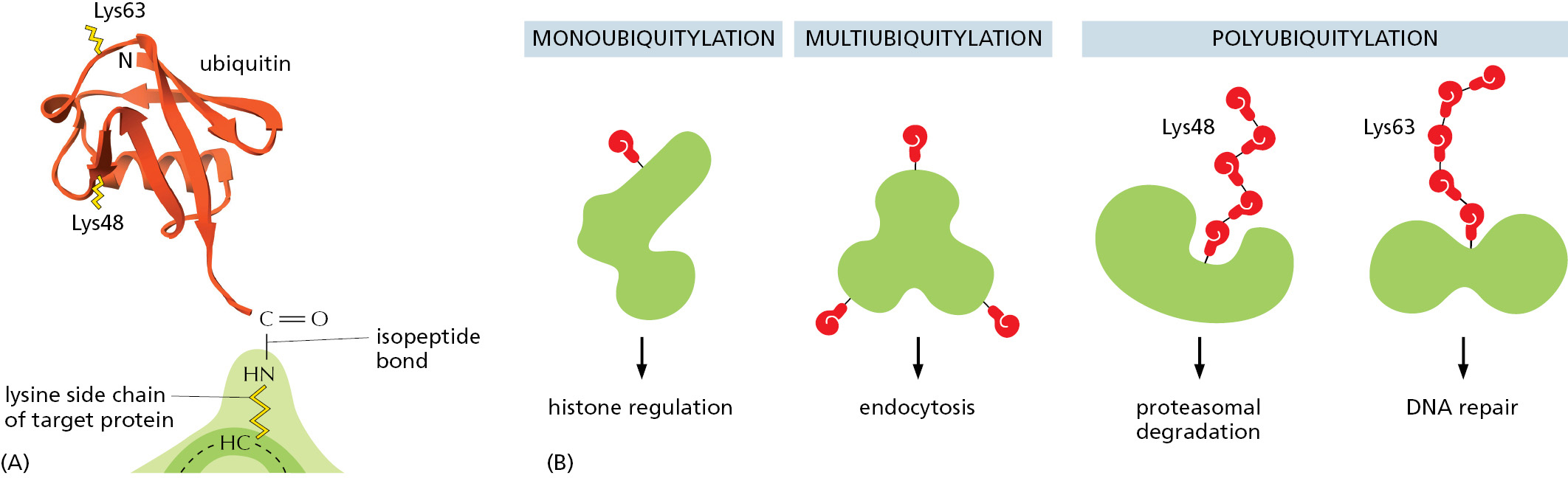
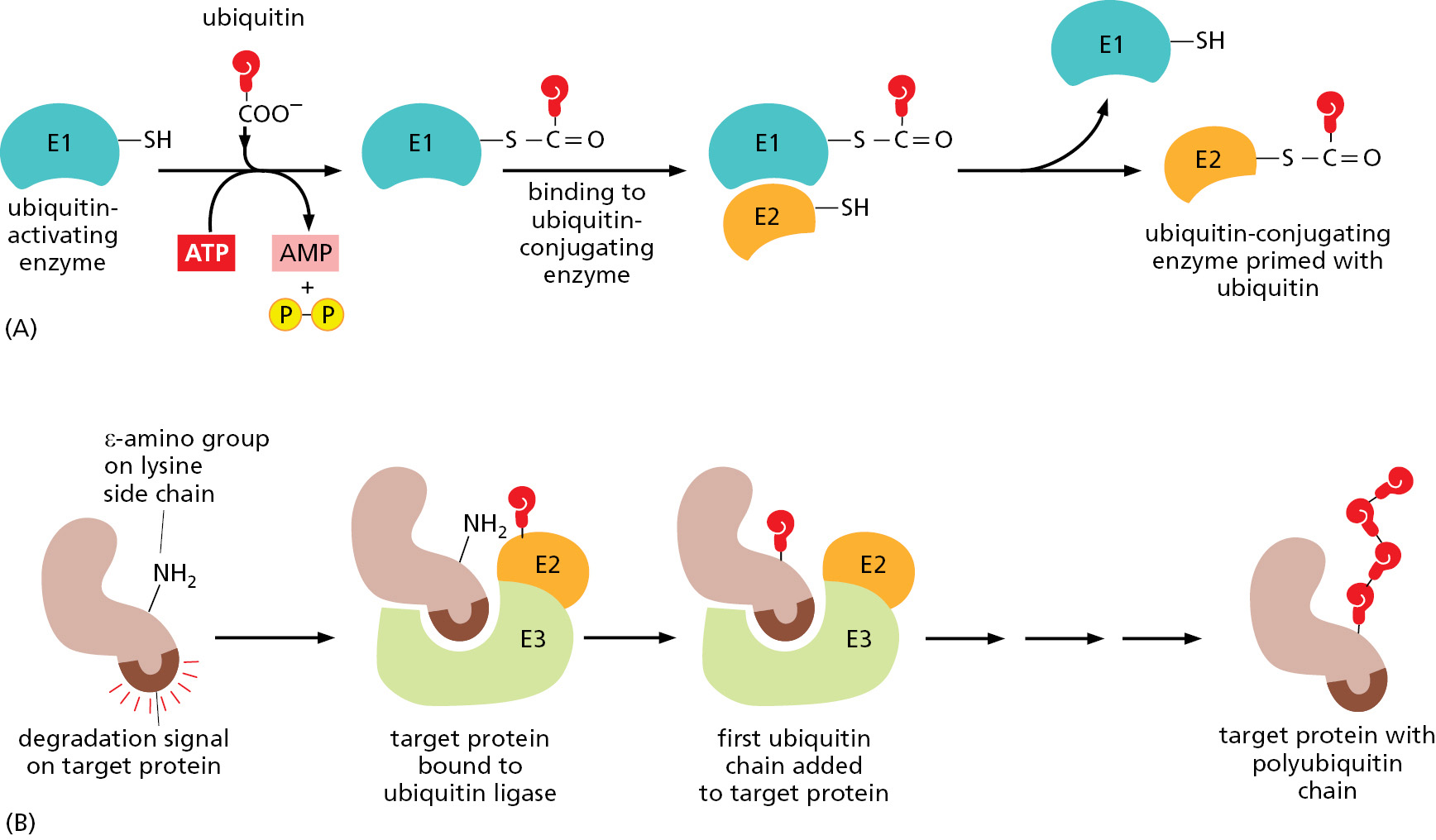
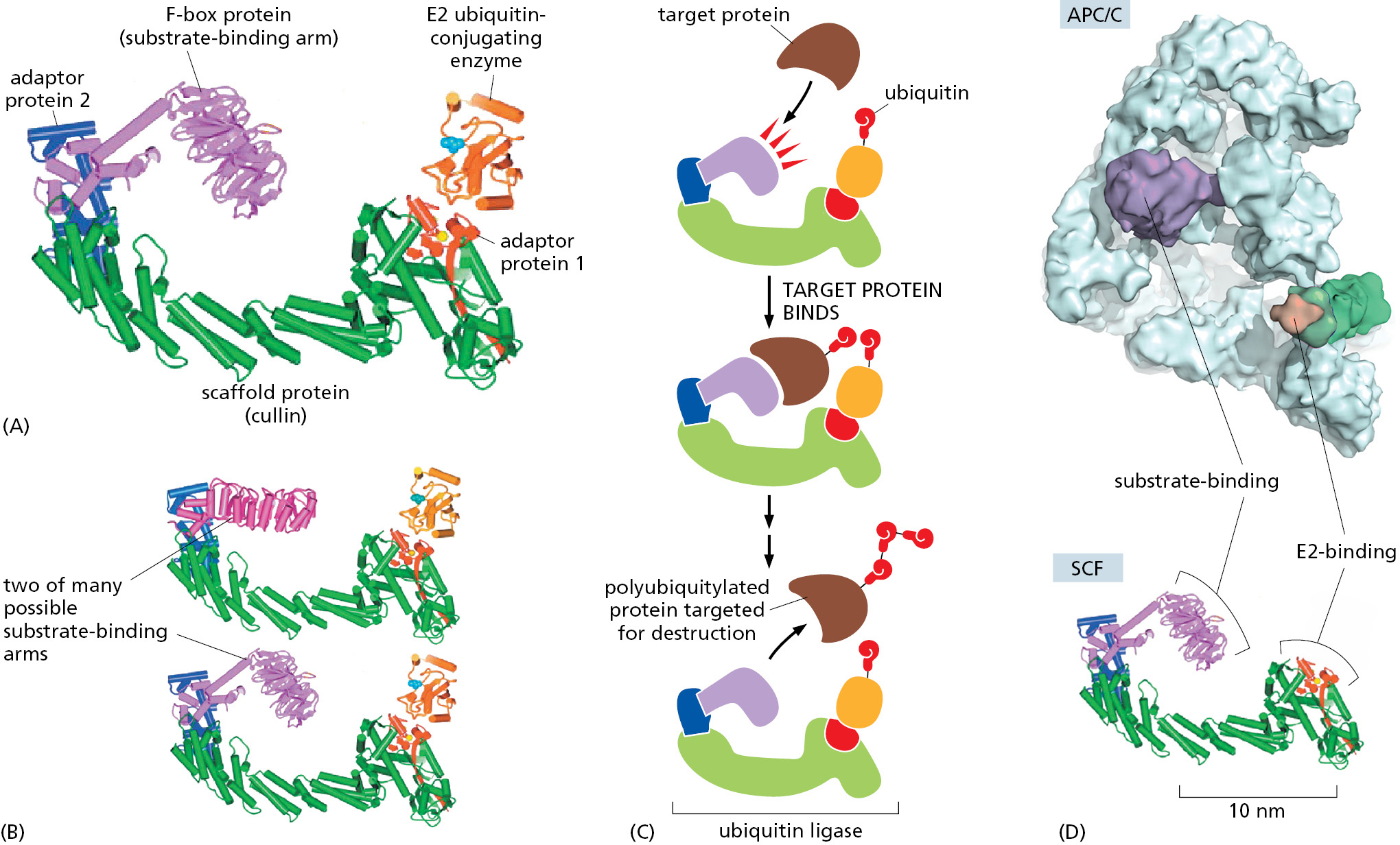
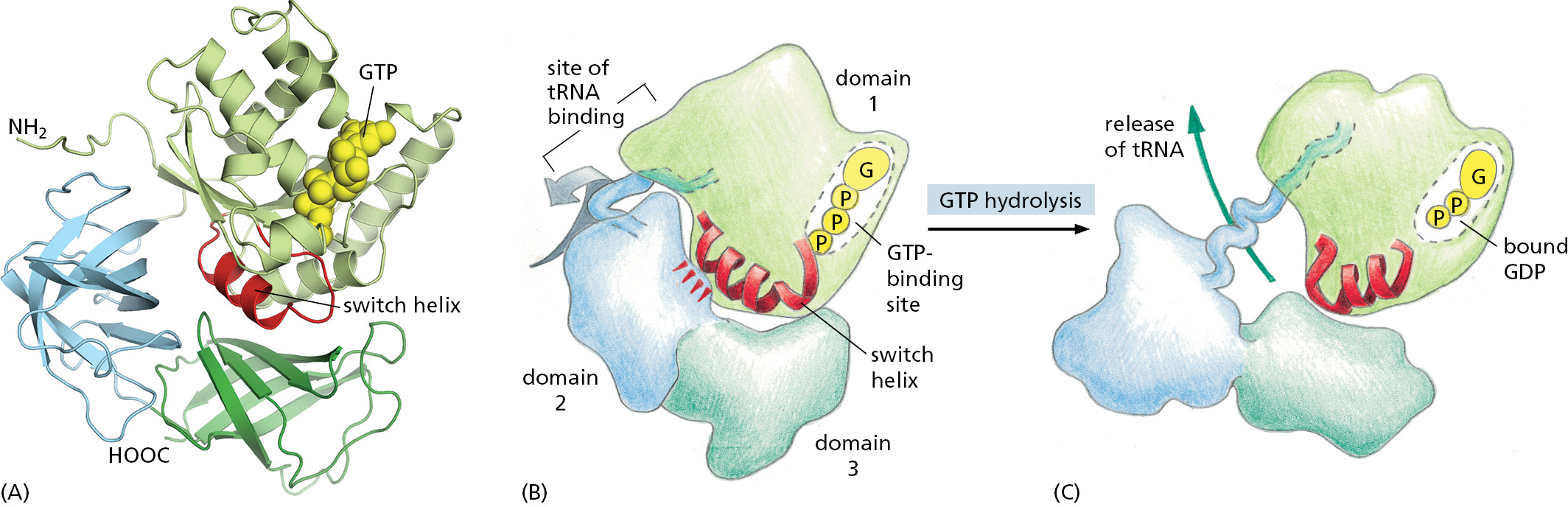
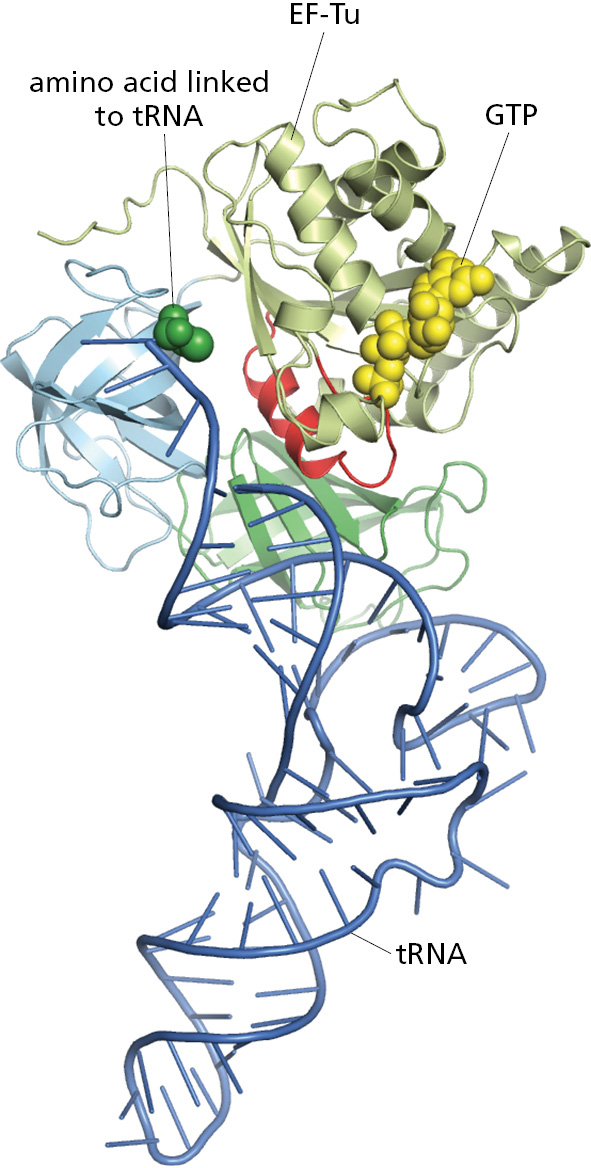
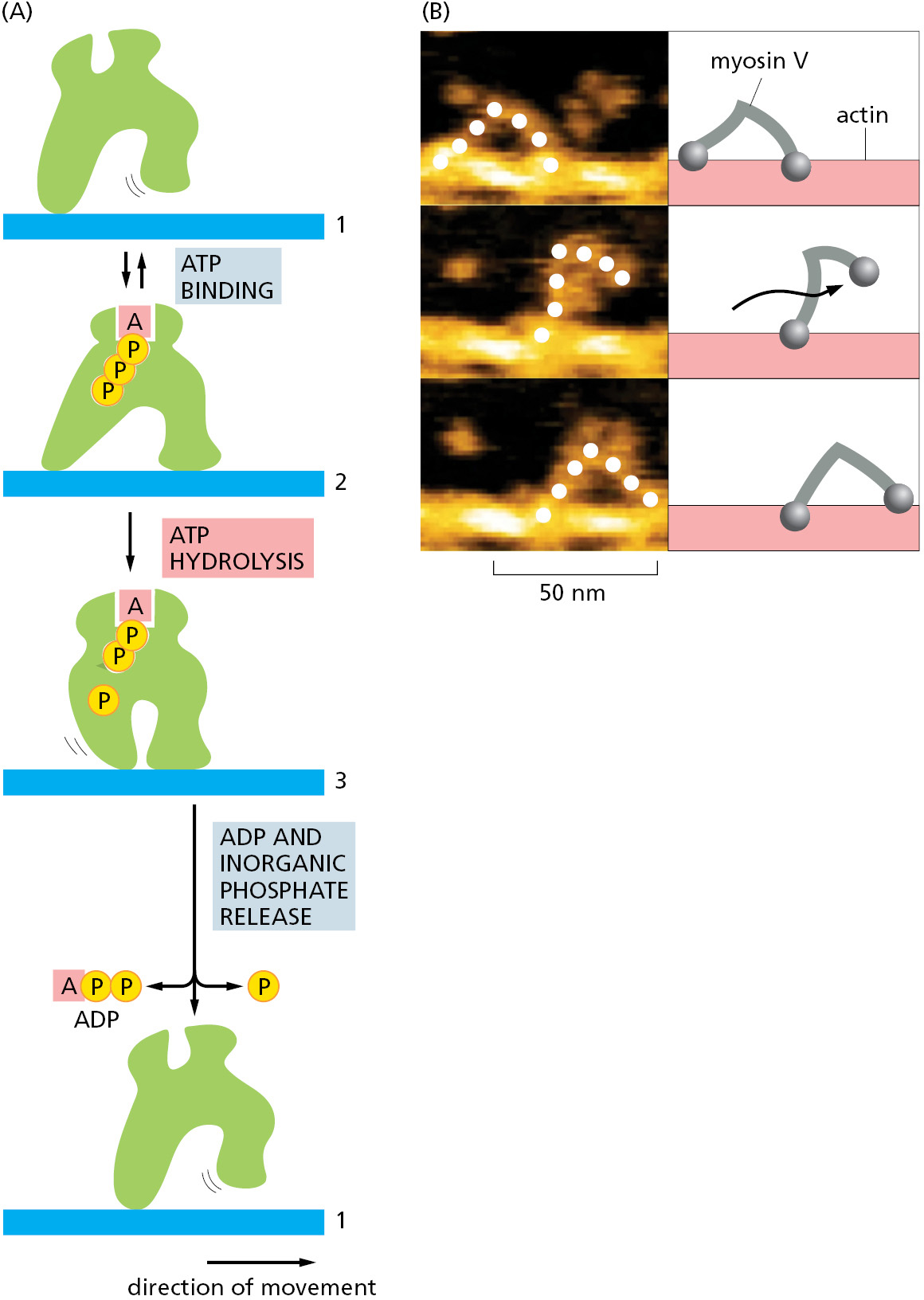
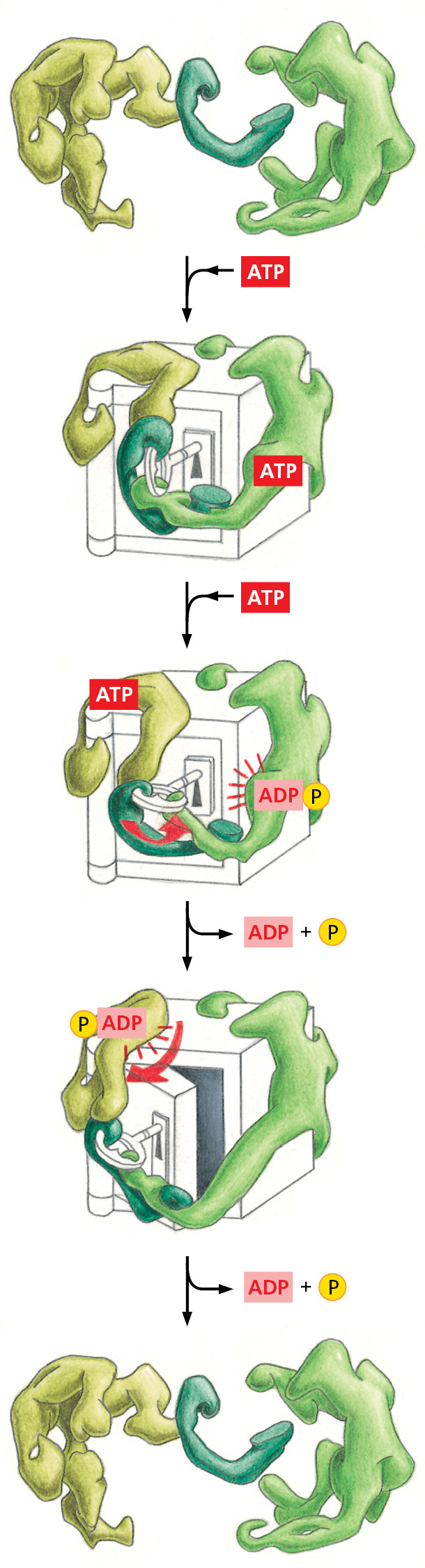
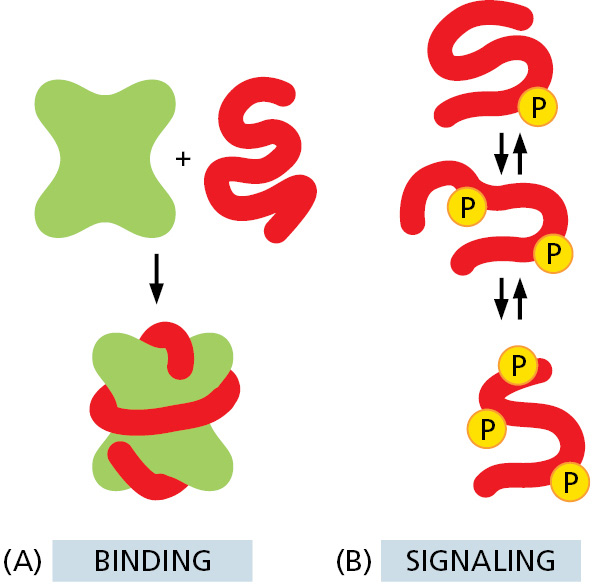
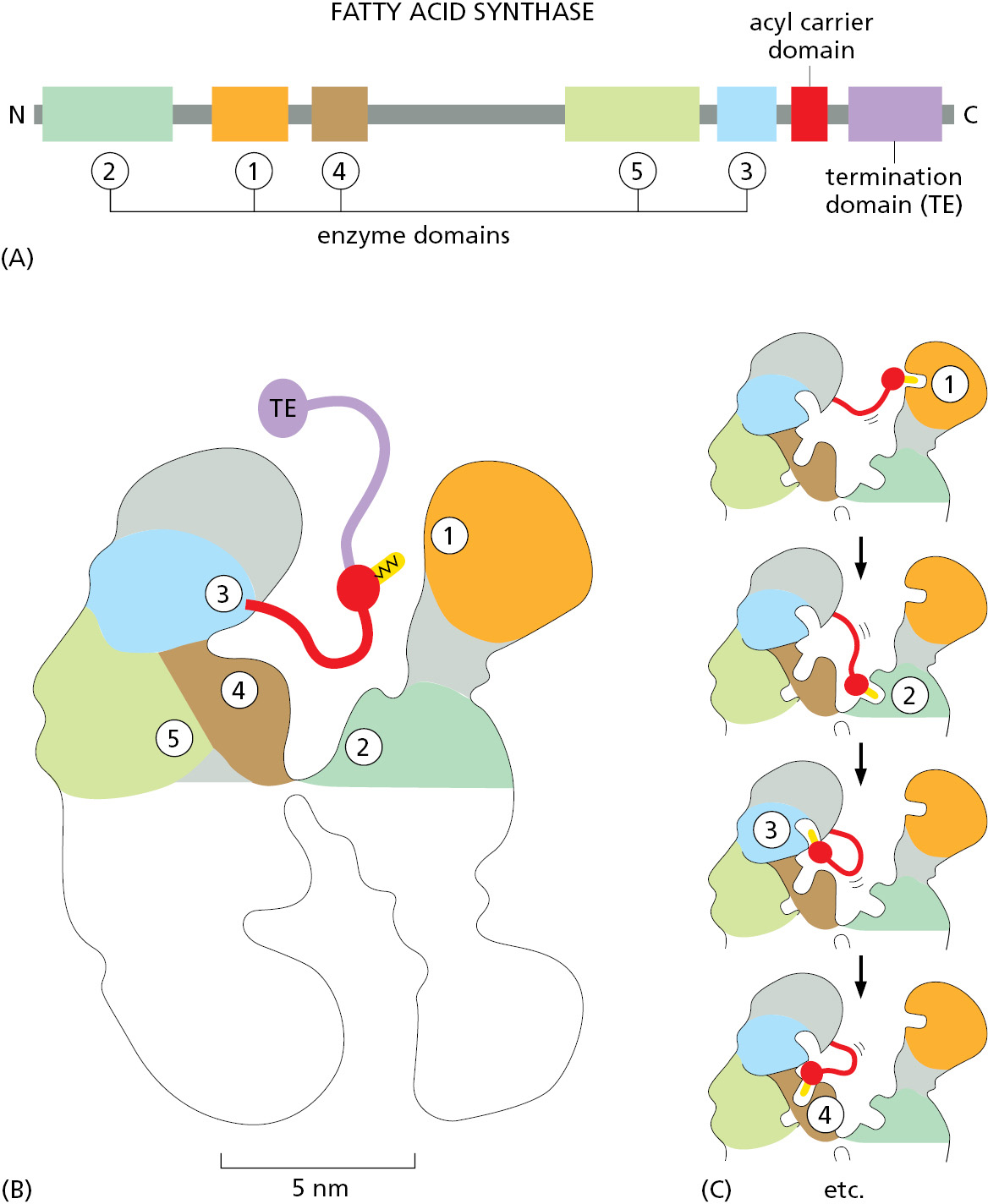
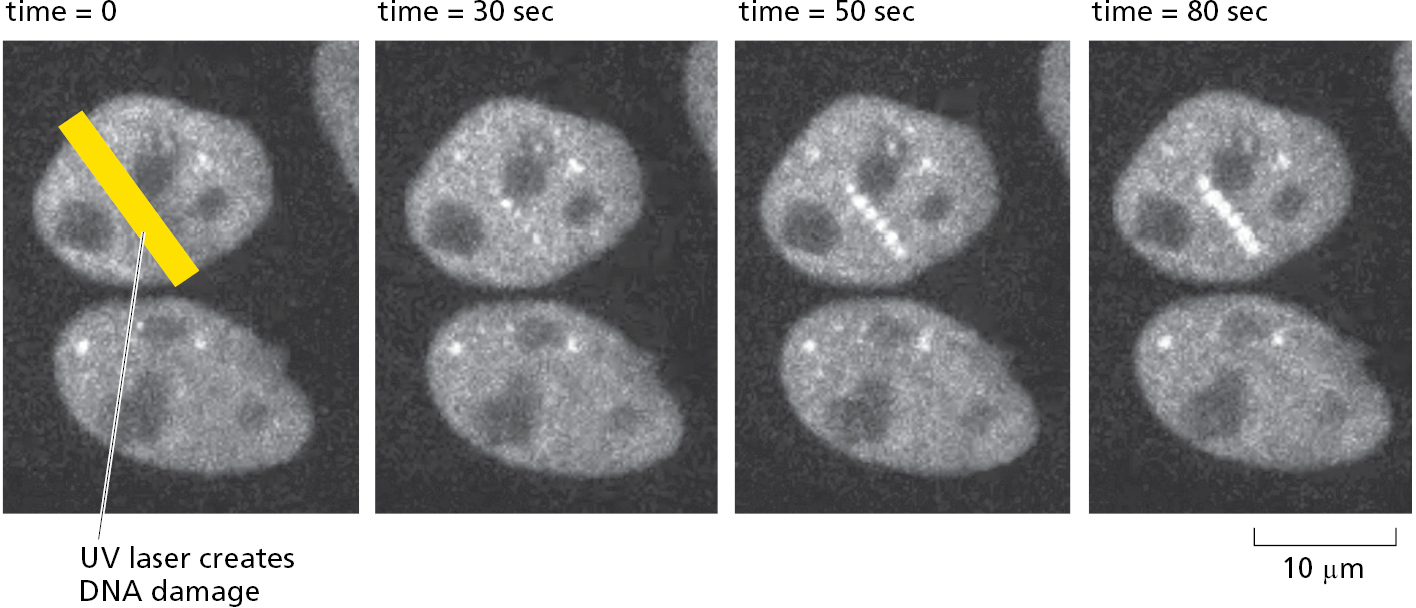
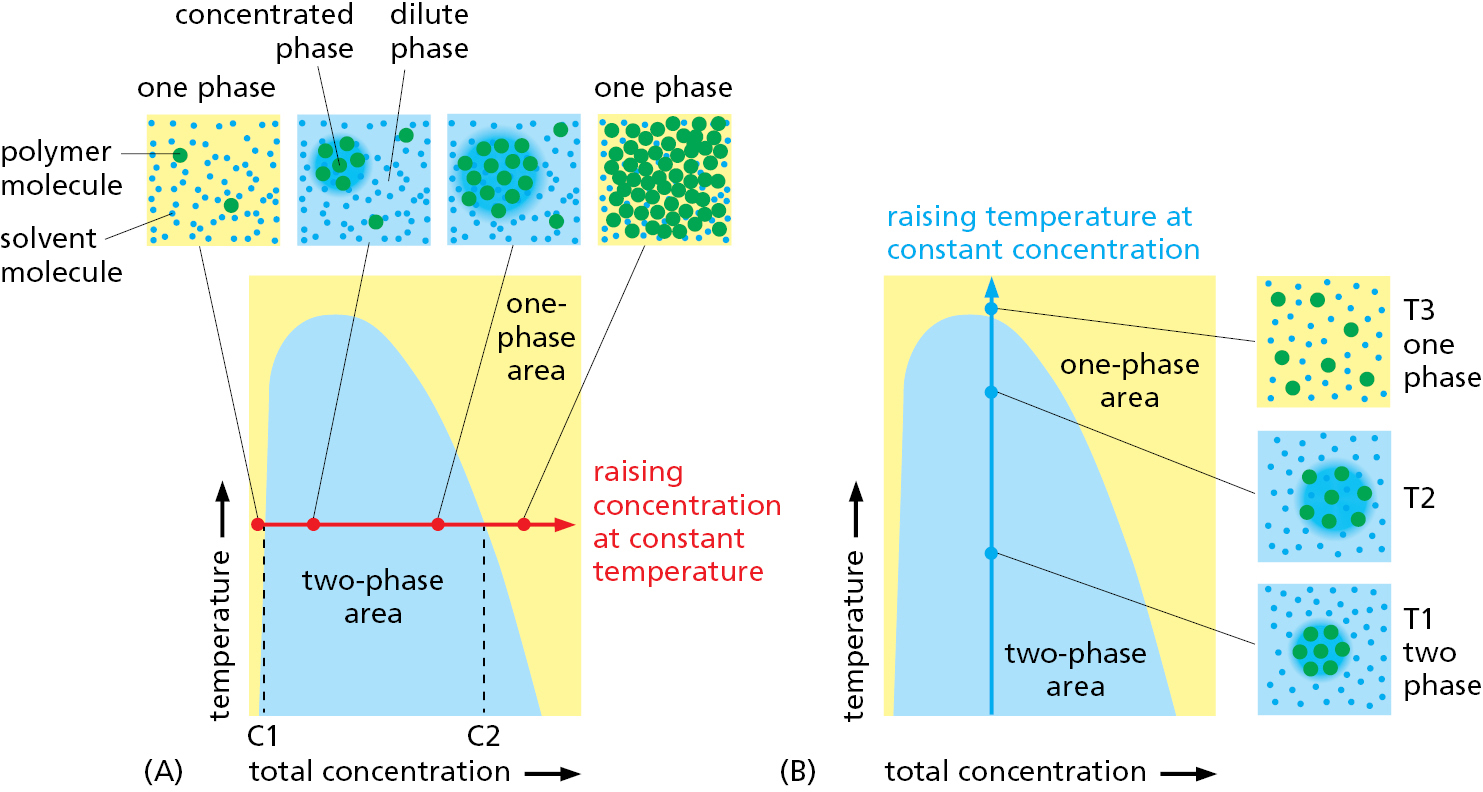
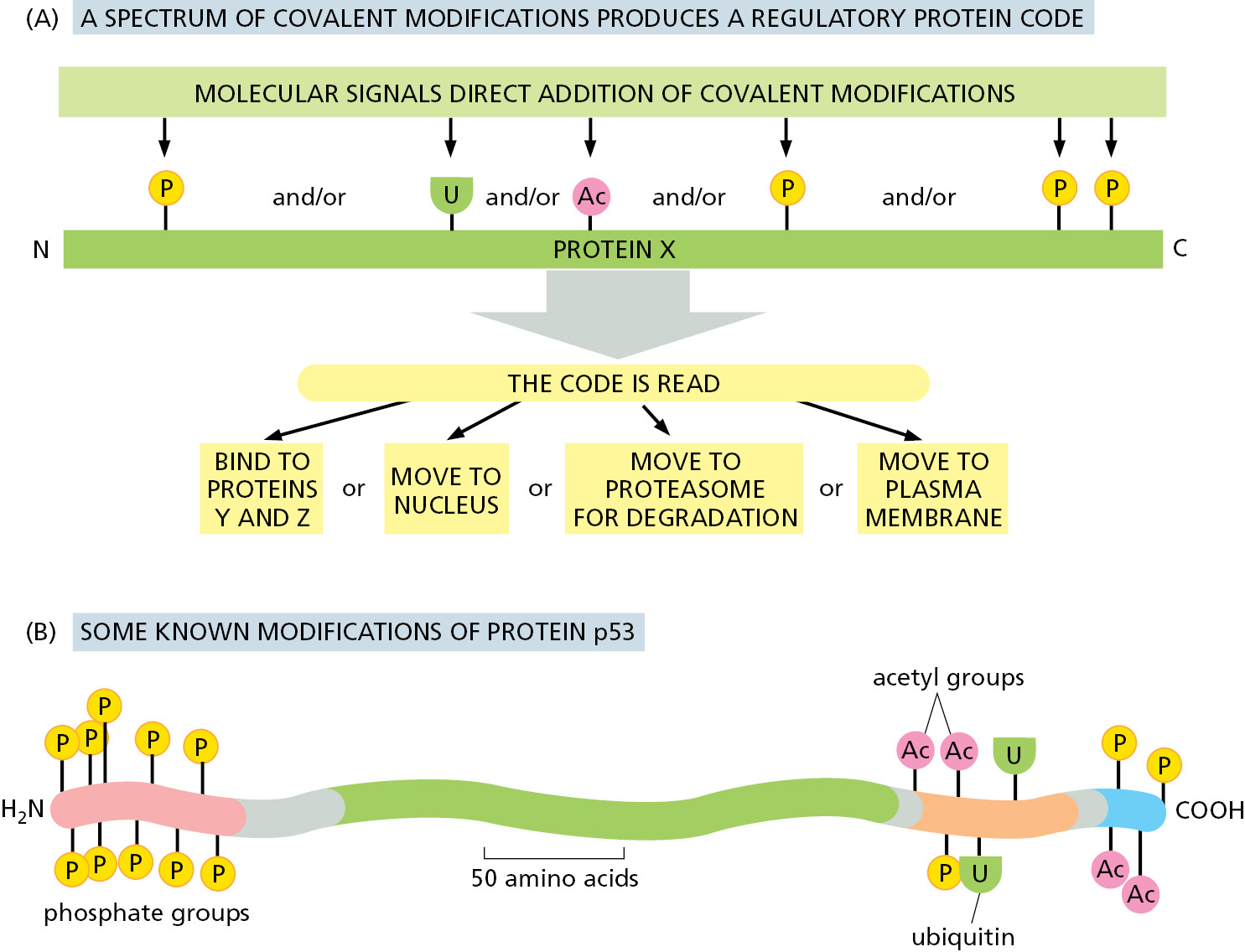
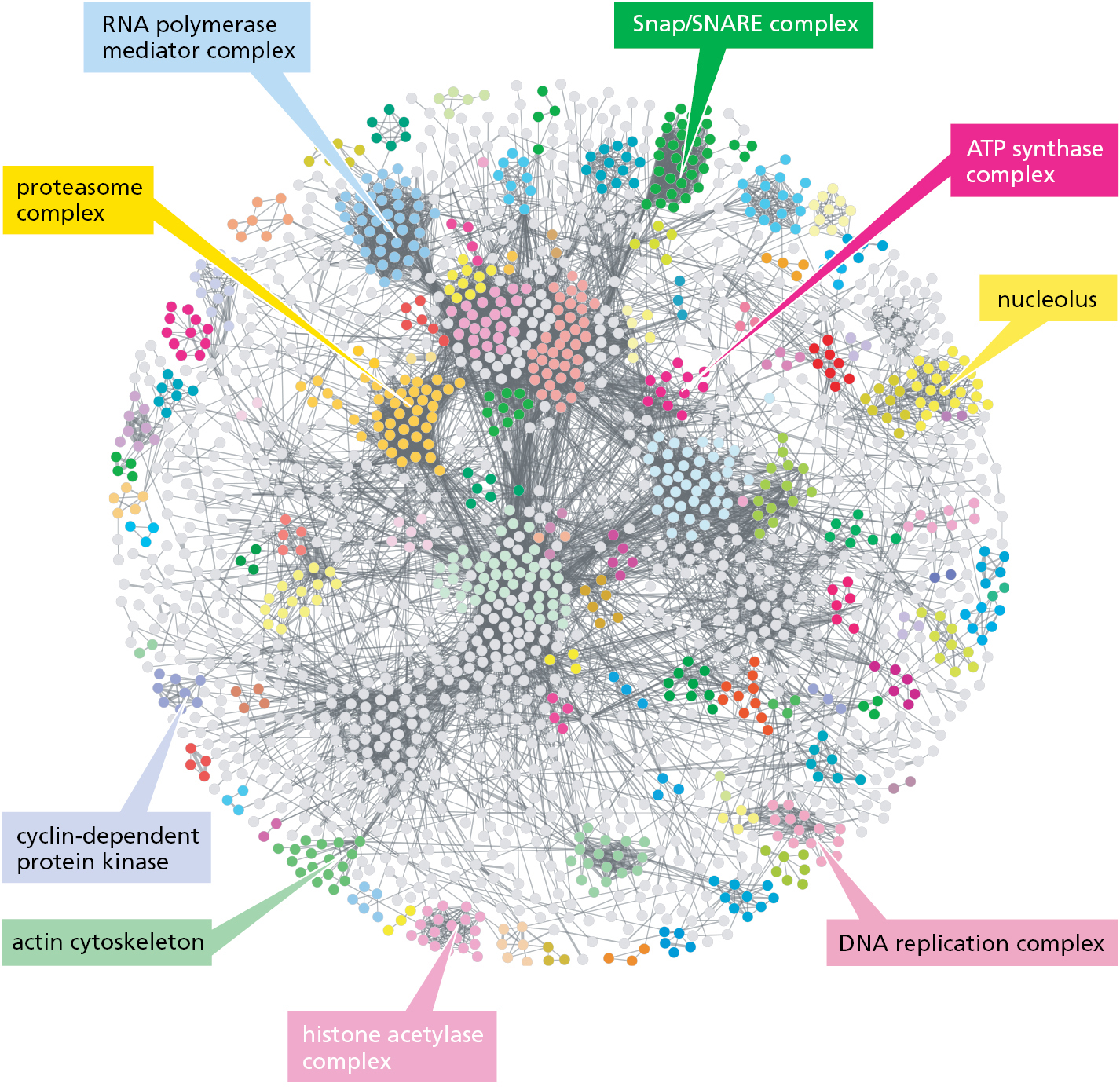
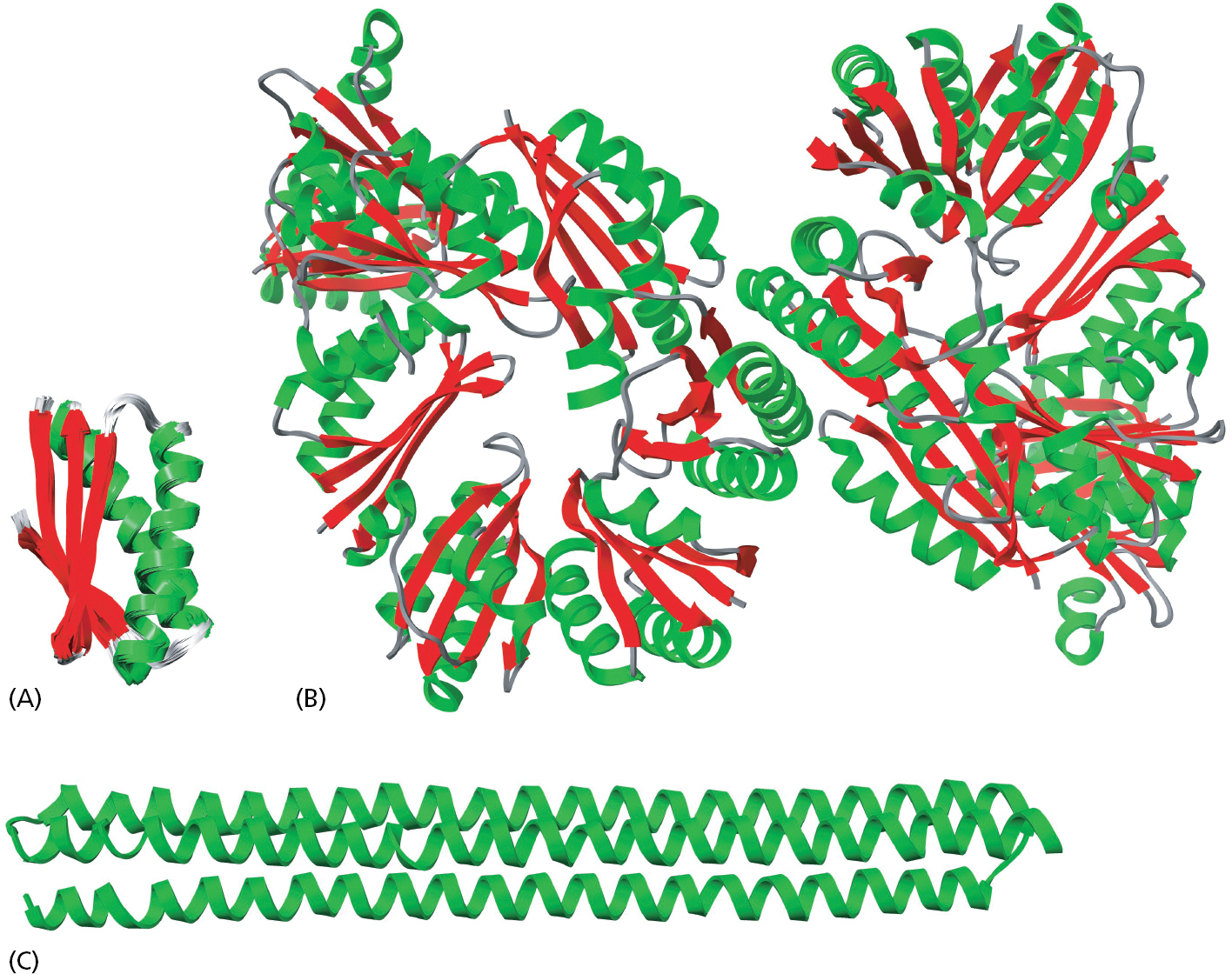
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}